RNA-Ribo Explorer user's manual
Table of Contents
Contact : translatome@lirmm.fr
Date : 2021-03-16
1 Quickstart
This program has been tested on the following operating systems: Linux (Ubuntu 20.04.2 LTS), and Windows 10.
Installing and lauching RRE
The easiest way:
- Download the jar file application named
RRE.jarfrom https://gite.lirmm.fr/rivals/RRE/-/releases. - Download also the jar file of the companion tool
FSTC.jarfrom the same site. - Click on the icon of the file
RRE.jarto launch its Graphical User Interface.
The Dashboard window should appear on your screen. You can then proceed with analyses. Read further from section 1.
Testing
We provide two datasets for loading into the RRE using the Graphical User Interface.
In the subdirectory data you should find two datasets with two files each (four files in total) as follows:
Each dataset has a file with a suffix _cov_table.txt
and another "twin" file with a suffix _orf_table.txt.
Attention: Both must be located in the same directory if you want to use them with RRE.
| Size in bytes | Filename |
|---|---|
| 5776459 | SRR1802148_cov_table.txt |
| 282843 | SRR1802148_orf_table.txt |
| 5147901 | SRR1802153_cov_table.txt |
| 285208 | SRR1802153_orf_table.txt |
In the File menu, use Import file and select always the _orf_table.txt of the dataset you want to import.
So if you want the dataset for the sample SRR1802153 you should import the file SRR1802153_orf_table.txt and no other.
For testing, import both datasets in the same way.
Installation from git repository
If you wish to recompile the program on your system, you can fetch the source code from the gitlab repository (URL: https://gite.lirmm.fr/rivals/RRE.git).
In Linux terminal:
git clone https://gite.lirmm.fr/rivals/RRE.git # get into the created directory cd RRE # compile with ant ant
This will create the jar file in the subdirectory named dist.
You may need to install the utility software ant from https://ant.apache.org/bindownload.cgi.
Launch RRE from terminal.
In a Linux terminal, type
RRE.jar
if you are in the directory where the jar file is, or if the jar file is placed in a directory that appears in your $PATH environment variable.
If you type
RRE.jar --help
it will print an help message and a description of the program, and then exit.
2 Overview
Let us start with the context and rationale for introducing RNA-Ribo Explorer (RRE for short).
Translation is a major step in the control of gene expression. Ribo-seq (RS) is a sequencing experiment that captures the fragments of an RNA that are covered by a ribosome during the translation process. The reads of a RS experiment are also termed Ribosome Protected Fragment or RPF for short. By aligning the sequence of read to all known RNA sequences of a reference database, one can deduce which part of RNA were undergoing translation in the studied cell. After this pre-processing step, the user may want to explore the data of multiple experiments, to compare them globally, or to inspect the RS profile of single RNAs. To our knowledge, an integrated tool for exploring such data interactively was missing. This is the reason why we developed RRE. Besides those features, RRE offers a data mining capacity where the user can design and perform queries on the RNA RS profiles of all target RNAs and RRE will automatically determine the subset of RNAs that fulfils the query conditions.
RRE is an interactive and flexible tool that enables the user to explore and analyse ribosome profiling data in a dynamic, visual, and easy manner. RRE is intended for the biologist for it allows her/him to design queries exactly matching his/her needs, to obtain the query results as selection or plots, and to examine graphically the results for any desired mRNA. The user can also set key parameters interactively, which will refine the results on-the-fly, giving her/him the possibility to test hypothesis dynamically. Furthermore, RRE does not require know-how in computational biology, nor a complex installation procedure, and should be as platform independent as the Java programming language is.
Terminology / Vocabulary
- RNA-seq: an NGS assay that reveals the presence and quantity of RNA in a biological sample at a given moment.
- Ribosome profiling: Next Generation Sequencing assay targeting messenger RNA (mRNA) to determine which mRNAs are being actively translated.
- Ribo-seq: short name of Ribosome profiling
- Coding region or CDS: portion of a gene's DNA or RNA that codes for protein. Also termed CDS for coding sequence.
- a RPF: Ribosome Protected Fragment: the piece of RNA sequence covered by the ribosome during translation and read by Ribo-seq. It is used as synonym of a Ribo-seq read.
- an ORF : Open Reading Frame: region of RNA sequence that can be translated into a peptide or a protein. It starts with a start codon (usually with an AUG codon) and ends with a stop codon.
- RPKM: Reads Per Kilobase per Million
- GFF format: generic feature format, or GFF, is a file format used for describing genes and other features of DNA, RNA and protein sequences.
Installation and requirements
Requirements: RRE should run on Linux, Windows, or MacOSX platforms. You should have JAVA working on your computer. This is usually the case on most computers nowadays. Otherwise you can get JAVA here.
Installation: The code is available on the gitlab repository. You just need to clone the repository, compile the source for your computer, and launch the main program. Details of the installation are explained on the wikipage of RRE .
3 Preparing datasets
RRE is an exploration software and it takes as input counts obtained by processing the reads. The processing is performed with a pipeline that consists in multiple steps (Figure 1). The first step basically maps the reads against a reference genome (or a reference transcriptome) for the chose species. This is typically done using external mapping tool (like BWA or CRAC).
It yields a SAM or BAM file.
The second step converts the SAM/BAM file (ie. the results of read mapping) into counts, with respect to known reference gene/RNA annotation. We provide a companion tool, called FSTC.jar for performing this step.
The third step is for downstream analysis where RRE comes into play. The user can load such counts data files into RRE to explore the data, select subsets of interesting RNAs, export information into tabular format, or produce graphics and figures.
For the second step, use FSTC.jar, the companion tool of RRE. It is also a JAVA application and is available from https://gite.lirmm.fr/rivals/RRE/-/releases.
It takes as inputs the SAM file and the Reference sequences annotation file, and produces the desired count data files.
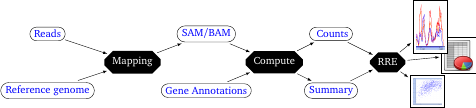
Figure 1: Logical overview of the read processing pipeline. RRE is used for interactive data mining in the final step (by the final user).
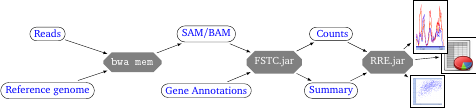
Figure 2: Same as in Figure 1 but with specific tools for each step.
How to compute the counts?
You can perform this using FSTC.jar, the companion tool of RRE. It is a JAVA application (FSTC.jar file) that you need to download independently.
- Inputs A SAM/BAM file for the mapping results. A FASTA and GFF for the references sequences and their annotations. Of course the identifiers of reference sequence must correspond in the FASTA and GFF files.
- Outputs
The application yields 3 files ending with special suffixes (by default):
- a coverage file ending with
_cov_table.txt, - a summary of annotation file for the ORF positions ending with
_orf_table.txt, - a log of the computation
reports.txt.
- a coverage file ending with
- Process It converts a SAM/BAM file into a coverage file. This coverage file stores for each transcripts the counts of mapped reads, with a record of reads' length. It also produces an ORF file, which contains the position of the main ORF within a transcript.
FSTC: description, usage, and example
FSTC is companion tool to RRE. It computes count data files from a SAM/BAM file and an annotation file for the chosen species. Each execution produces two output files (with specific suffixes in their names: _cov_table.txt and _orf_table.txt).
- Executing
FSTC.jar
In your terminal, if you are in the directory where the jar file is, or if the jar file is placed in a directory that appears in your
$PATHenvironment variable, type:FSTC.jar --help
it will print an help message and a description of the program, and then exit.
The usage is printed if you type
FSTC.jar
If
FSTC.jaris not found by your terminal, but you know it is in the directory, sayprograms, then you can still type:java -jar programs/FSTC.jar
- Usage with options
Usage: FSTC.jar --GFF your_annotation_file.gff --output-prefix result_prefix --SAM your_sam_file.sam OR FSTC.jar --GFF your_annotation_file.gff --output-prefix result_prefix --SAM your_bam_file.bam Mandatory parameters: --GFF /your/annotation/file.gff --output-prefix /your/output/prefix --SAM /your/input/reads.sam Optional parameters: --allSources take all contigs in annotations. default: only mRNA with BestRefSeq are considered. --addDuplicatedGene keep duplicated genes. default: they are removed. --addPartialGene keep partial genes. default: they are removed. --forgetFlag do not look at SAM flags. default: they are taken into account. - Example with input and output files.
In a given directory, you have:
- a bam file (the output of a mapping tool)
SRR493747_transcriptome.bam - a GFF file with gene/mRNA annotations for the chosen genome or transcriptome:
GCF_000001405.30_GRCh38.p4_genomic.gfffor the Human genome
Moreover, we assume that
FSTC.jarcan be found and executed on your system (In Linux, this means that it is in a directory of your$PATHenvironment variable). Then, in a terminal you can type the following command:FSTC.jar --GFF GCF_000001405.30_GRCh38.p4_genomic.gff --output-prefix SRR493747 --SAM SRR493747_transcriptome.bam
it should print
reading annotations : done. (19060 selected genes) ---------- Annotations were compiled. ---------- Reading SAM/BAM is done. (1906397 reads) done.
and output 6 text files (whose names start with the string located after option
--output-prefix):SRR493747_annotations_genes_table.txt SRR493747_annotations_summary.txt SRR493747_counts_report.txt SRR493747_noRead_genes.txt SRR493747_cov_table.txt SRR493747_orf_table.txt
The two last files (
SRR493747_cov_table.txtandSRR493747_orf_table.txt) are required by RRE. They need to be located together in the same directory. These are text files, so do not open and save them with Word, Openoffice, or any similar text processing software.For testing FSTC with this example, you can fetch
- the BAM file
SRR493747_transcriptome.bamat
https://seafile.lirmm.fr/f/ab296da765cf46f086c7/?dl=1 - and the GFF file
GCF_000001405.30_GRCh38.p4_genomic.gff
from https://seafile.lirmm.fr/f/ac333afda6274f1fb52b/?dl=1 .
To check what it outputs, you can fetch a whole directory (name
res_ori_bam) with the expected output files from https://seafile.lirmm.fr/d/b7a43d38fd94423985db/. - a bam file (the output of a mapping tool)
4 Loading data in RRE, parameters, saving a session
RRE can help you exploring, mining, or comparing multiple datasets at once. Datasets can be of two types: RNA-seq or RS. These datasets are loaded individually into RRE. Once you have explored some datasets with RRE, but have not finished, you can save the current session into an external file. This session file can then be reloaded later to carry on working with the same data. Finally, you can control the Session settings and the attributes relative to each dataset.
Dashboard
A view of the Dashboard is shown in Figure 3. When launching RRE, the dashboard appears without any dataset (just the logo of our lab). In this figure, four RS datasets were previously loaded and theirs attributes set. For instance, the shift of the first dataset has been set to 12 (while the original, default value is zero).
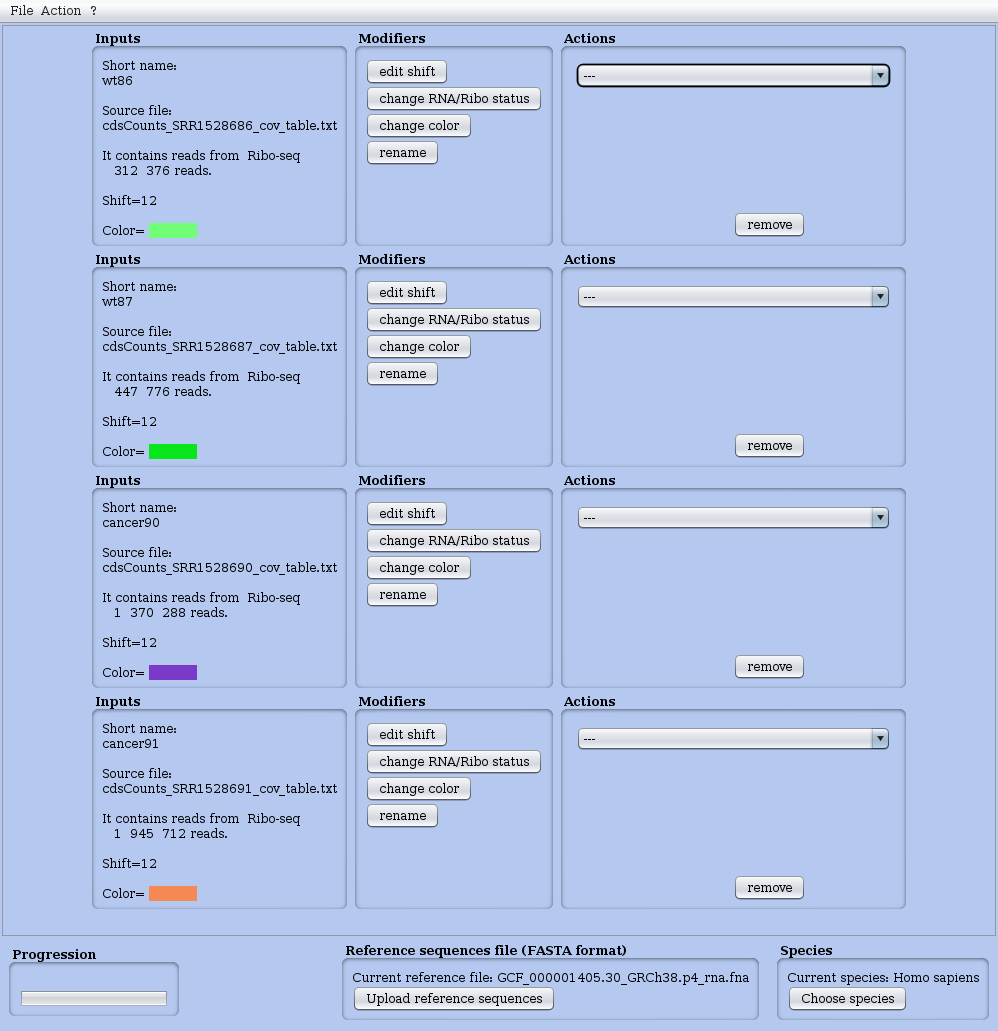
Figure 3: Dashboard (main window) of RRE with four datasets already loaded. Main menu upper left corner. In the window, all loaded datasets are presented one per line.
Loading count files
For one dataset you must have two files:
- a coverage file ending with
_cov_table.txt, - a summary of annotation file for the ORF positions ending with
_orf_table.txt,
They must be in the same directory.
Example:
Assume you have two datasets called A and B. Then, you must have the files
- files
A_cov_table.txtandA_orf_table.txt, - files
B_cov_table.txtandB_orf_table.txt.
To import them, choose in the main menu File the command Import data. A dialog box open: select the coverage file for example choosing A_cov_table.txt will upload both count file for dataset A.
Then a line appears in the Dashboard to represent this dataset. This line has three columns (see Figure 3).
- Inputs: a description of the dataset and its attributes
- Modifiers: four buttons to modify those attributes (see Figure 4)
- Actions: a menu to launch analyses
Attributes of a dataset
The following attributes are set or asked for each uploaded dataset (file).
- Offset (or shift) (specific feature for Ribo-seq). The tool provides a visual application to fix the appropriate offset. It can also determine an appropriate value for the offset by globally examining the data with respect to annotated ORF.
- Short name: It will be used in graphs and tables produced by RRE.
- Colour: It is used in graphs to visualise data from this dataset and it eases comparing multiple datasets.
- RNA-seq or RS status: The status is a Boolean information indicating the type of this dataset: either set to RS or to RNA-seq. If the status is RS, the application will discard too long or too short reads. With another status, reads of any length are considered.

Figure 4: Zoom in the Dashboard (main window) of the modifiers of a dataset. Just after loading a dataset, you must edit the shift to define the offset for positioning the codon undergoing translation from the read position.
Session settings
RRE has some general settings or parameters that control its behaviour independently of the datasets. To set these parameters use in the main menu File the command Parameters.
A window opens up and displays the general parameters in a table (Figure 5).
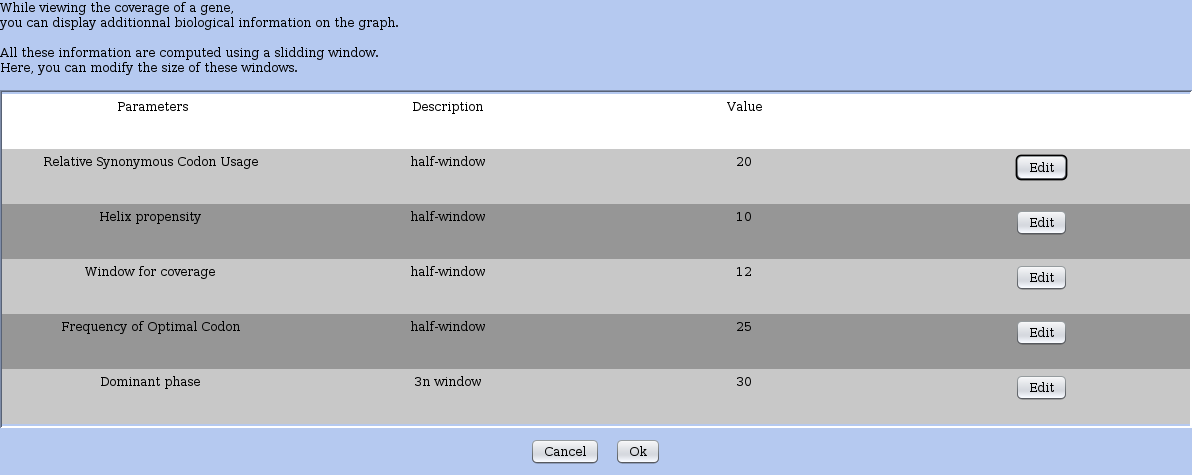
Figure 5: Session settings: window to control the general parameters of RRE. Parameters description, current value are shown in a table and an Edit button allows you to change their value. All parameters set the size of a sequence window that is taken into account to compute various statistics. When the window size needs to be even, one sets the half-window size. Regarding the determination of the dominant phase: the window size is constrained to be a multiple of 3.
| Parameters | Description |
|---|---|
| Window for coverage | This relates to the sliding window option when viewing the coverage of an RNA. Averaging coverage is performed over this window length at each sequence position. |
| Relative Synonymous Codon Usage (RSCU) | Global statistics measuring the bias of codon usage within the RNA coding sequence. See the RSCU page and Paulet et al, DNA Research 2017 paper. |
| Frequency of Optimal Codon (FOP) | Another statistics measuring the bias of codon usage through the preference of optimal codons. |
| Helix propensity | Statistic measuring the propensity of the sequence in the window to form helices. |
| Dominant phase | Used for determining which phase (0, +1, +2) likely contains an ORF. |
Remark: Only the Window for coverage modifies the behaviour of RRE. The other parameters are for computing and viewing additional biological information, but do not alter the way queries are handled or the RS profile visualisation.
Reference for the RSCU using RS data:
Ribo-seq enlightens Codon Usage Bias
D. Paulet, A. David, E. Rivals
DNA Research, dsw062. doi: 10.1093/dnares/dsw062, 2017.
Saving a session
Often, you may want to re-explore later the data on which you have worked today. RRE allows you to save the current working session on a disk on file. You may then later reload this session by reloading the data file. The file contains the proper data (which is counts of read at each position for all RNAs), but also the parameters, the queries and the selection that you have made until now.
To do this in the main File menu use Save session and Load session respectively.
5 Control analyses
Distribution of read length
A usual control for RS data is to inspect the Distribution of read length in the dataset. In RS protocol, the fragments are selected by length to fit the expected length covered by a single ribosome during translation (usually around 28 nucleotides). The reads may slightly depart from this length and viewing the distribution is a way to spot for "outliers", that is for reads with "abnormal" lengths.
The Actions menu choose draw the distribution of read lengths to obtain this plot – as shown in Figure 6.
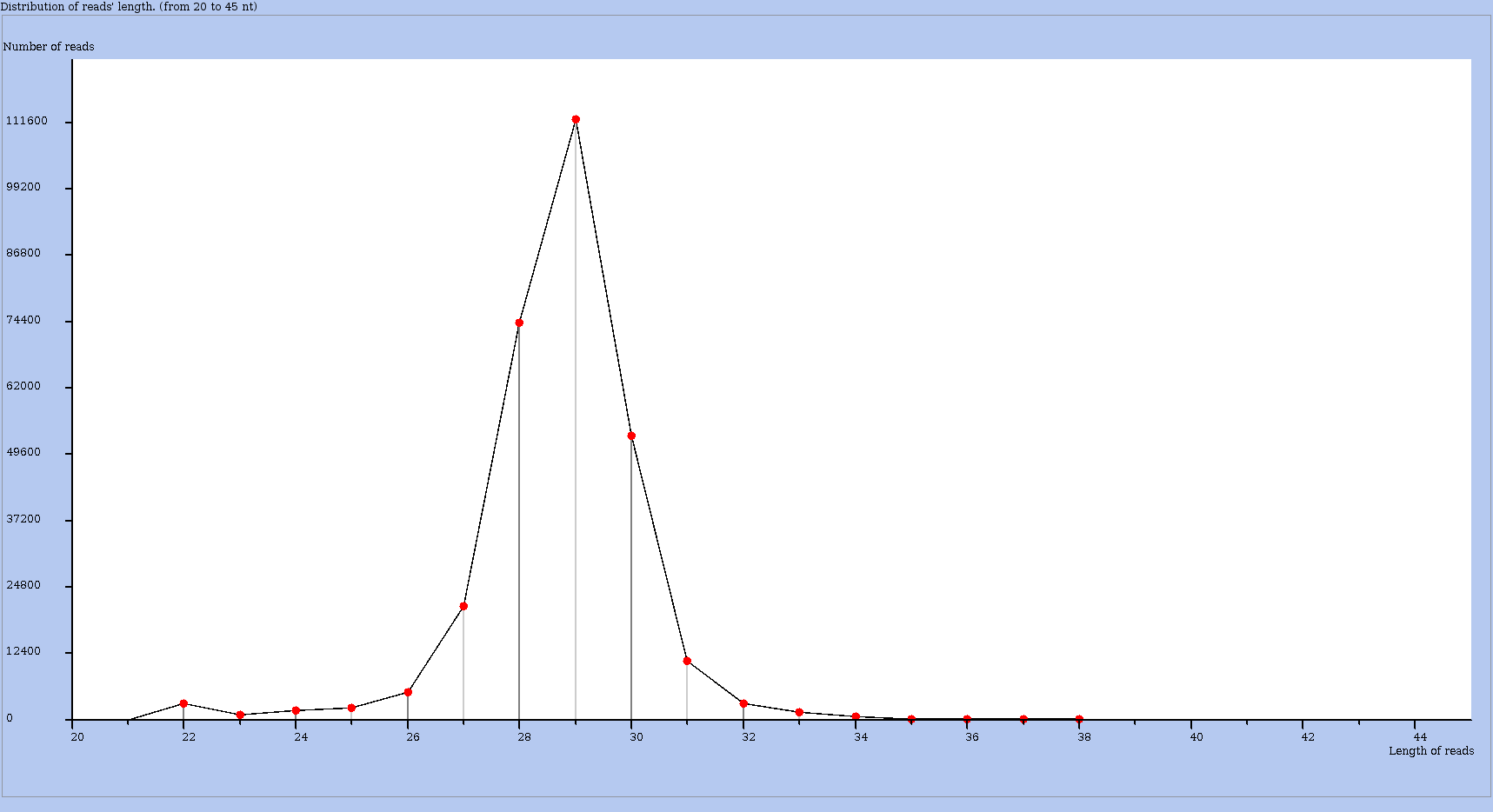
Figure 6: Distribution of read lengths for a chosen dataset.
Overall distributions of mapped reads in coding phases.
Take a look at the proportion of reads that were mapped in different coding phases. In the main Analyses menu choose Distribution of reads in phases to compute a summary presented in a table where you can compare the situation of all datasets simultaneously (see Figure 7).
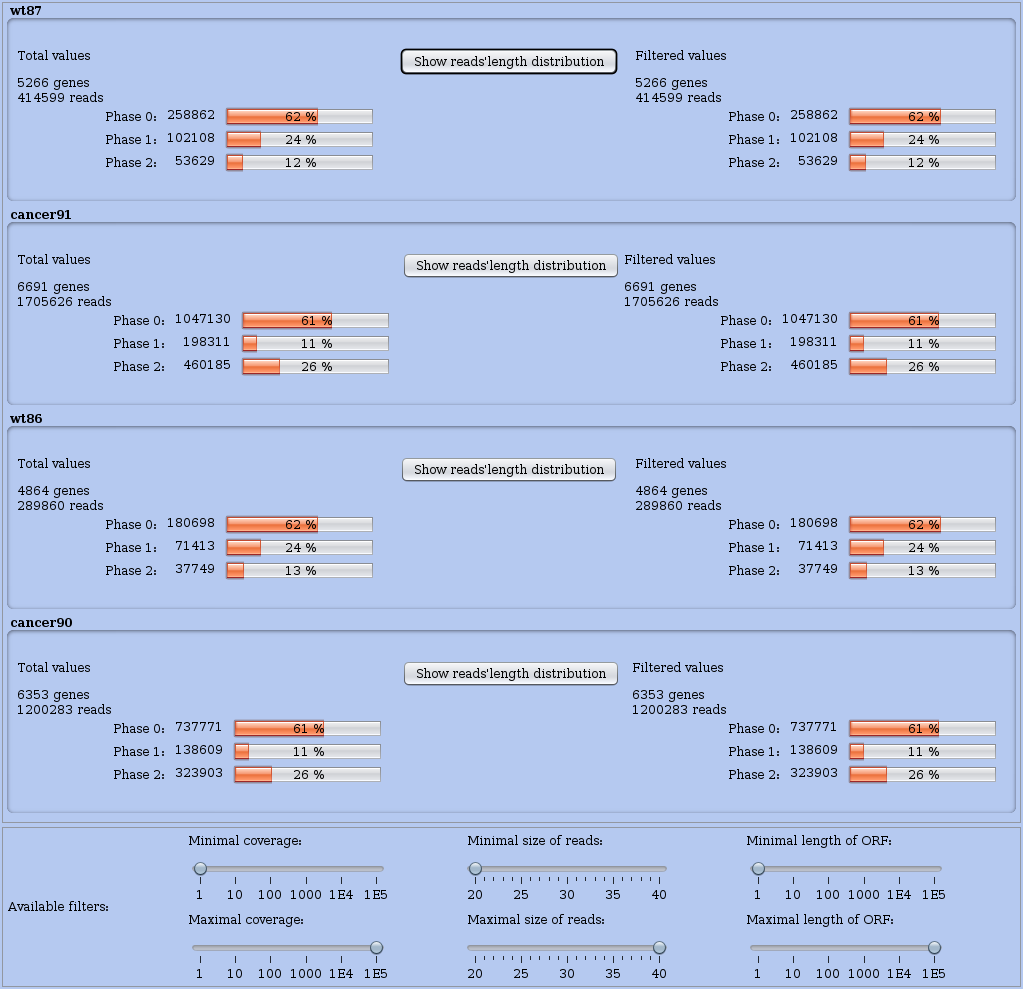
Figure 7: Distribution of read locations across coding phases in RNAs.
Each dataset is summarised on one line. Numerous information are detailed.
At the foot of this window, you may set thresholds or ranges using three panels one per feature. From left to right, each panel allows you to set the range of:
- the total coverage of each RNA
- the sizes of reads taken into account
- the length of ORF in each RNA.
Each dataset line comprises the summary for unfiltered data (on the left) and the same summary but for filtered data (on the right). Using the two cursors in each panel, you set the minimum and maximum values for each range, and the filtered summary for each dataset is updated immediately. That way you can control whether a subset of data is more appropriate for subsequent analyses.
Overall distributions of mapped reads across RNA regions (ORF and UTRs)
It is also interesting to examine the proportion of reads that were mapped in different kind of annotated regions: 3' UTR, main ORF, 5' UTR. In the main Analyses menu choose Distribution between UTRs and ORF to compute a summary presented in a table where you can compare the situation of all datasets simultaneously (see Figure 8).
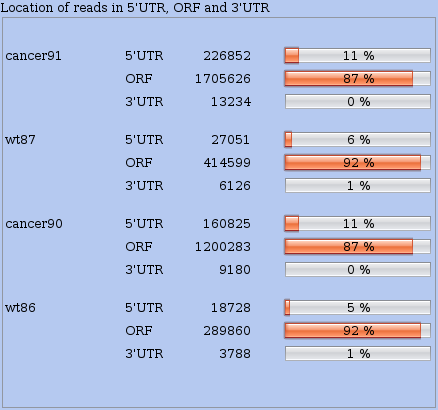
Figure 8: Distribution of read locations across annotated regions of all RNAs (3' UTR, main ORF, 5' UTR).
6 Visualising the profile of an mRNA
You can get the view of the RNA-seq or RS profile of an RNA by different means. Either directly from the Actions block of a dataset using draw one RNA or draw one RNA in several conditions. Or indirectly after analysing globally one dataset and producing a plot where each RNA is drawn as a dot. Then clicking on the dot opens up a contextual window, which allows you to get the profile view for this RNA. An example of profile view is shown in Figure 9 with coverage in absolute numbers or in Figure 10 with the sliding window option on.
The coding region of the main ORF is materialised by an horizontal blue bar below the profile along the X-axis.
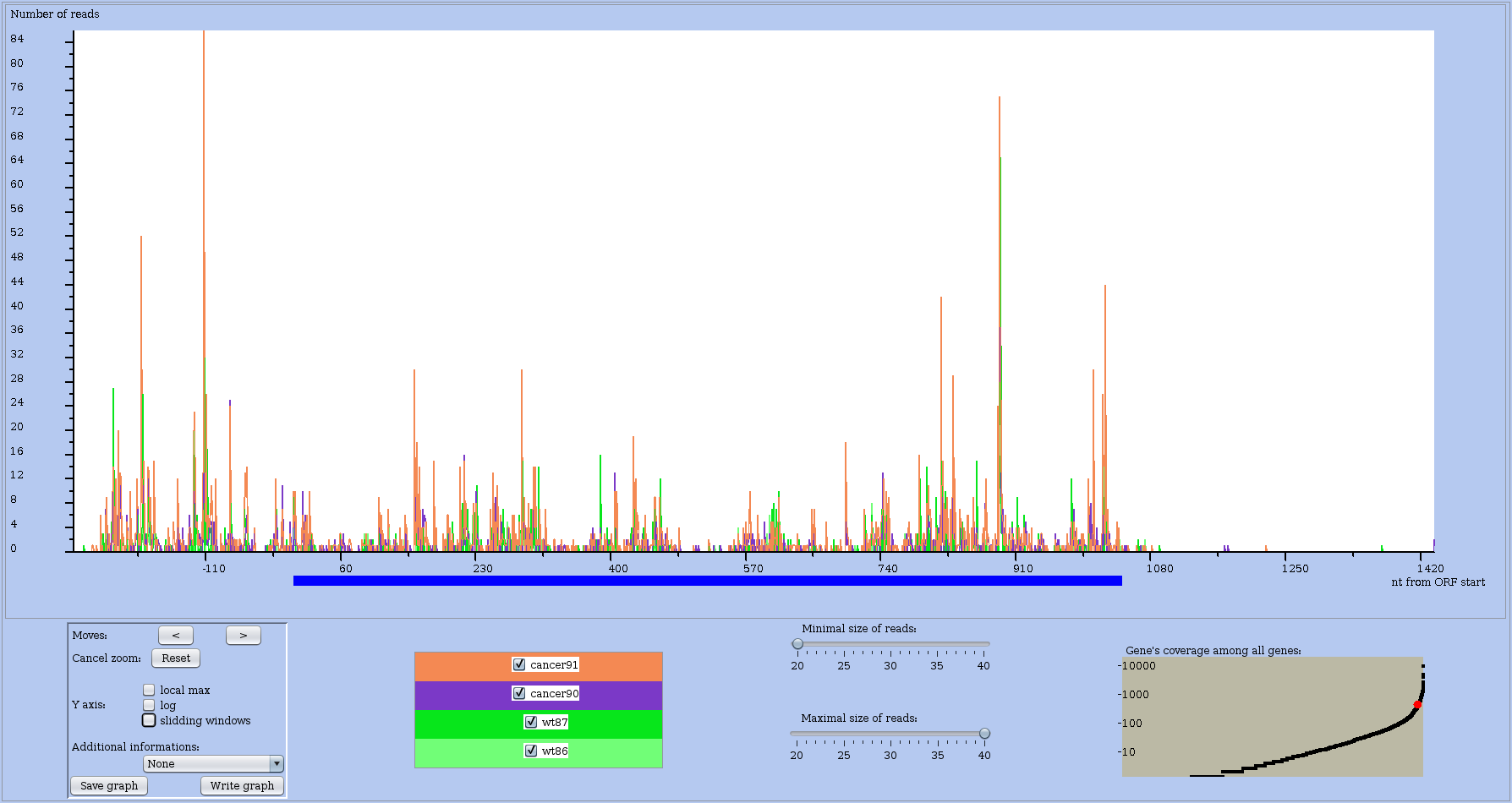
Figure 9: Coverage profile of a chosen RNA in raw form. Note that the RNA name or identifier is shown in the window frame, which is not shown here.
Nucleotidic positions: Current official annotations consider that there is one main ORF for each RNA. Usually it is the longest ORF among all. RRE arbitrarily (also) considers the translation start site of this main ORF as the reference position for the plot.
In the profile view, nucleotides are numbered according the translation start site. Left of it (that is towards the 5' end) nucleotide have a negative position, while right of it (towards the 3' end) positions are positive integers.
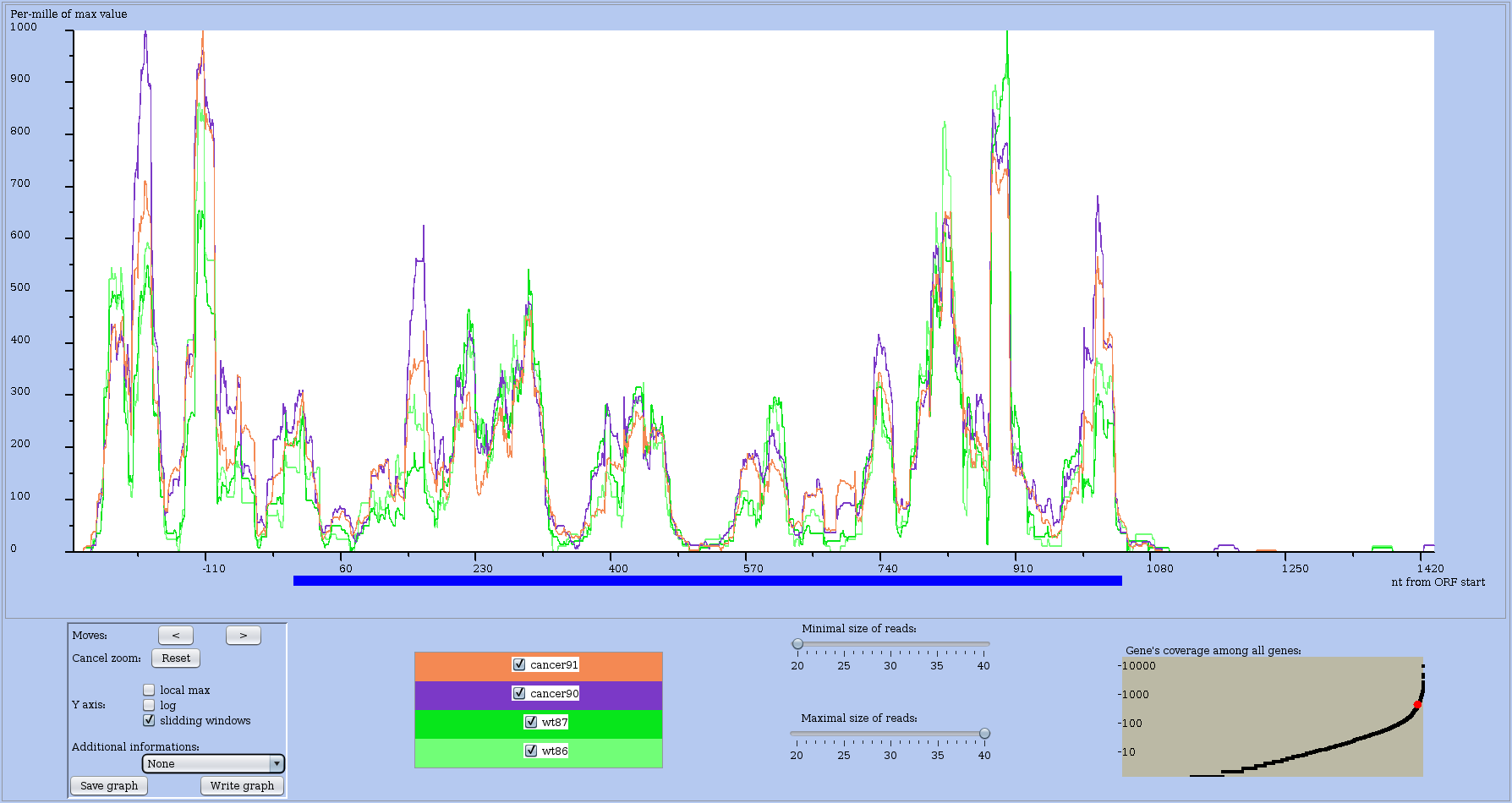
Figure 10: Coverage profile of a chosen RNA with sliding window option on. Same RNA as in previous figure.
Several options or control buttons allows you to change the profile view.
Left control panel (Figure 11)
First you can zoom by selecting a region of the plot using the left mouse button and dragging over the desired region. The profile view will be updated right away. To reset the zoom to 1x, just on the Cancel zoom reset button.
In the lower left corner, the panel comprises a log button and a sliding window button that let you trigger the option of a logarithmic Y-axis or the computation of the coverage over a sliding window. Currently the local max button has no effect. Thanks to the < or > buttons you can move left or right the view (only after zooming in).
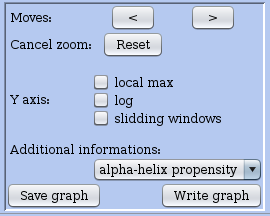
Figure 11: Left control panel on the profile view of an RNA.
This panel allows you to plot additional biological information regarding codon usage bias (FOP and RSCU), structural elements in the sequence (alpha helix propensity), a comparison of possible coding phases (zone per phase). Choosing one of them from the rolling menu will add a curve onto the plot to show the variation for the corresponding information.
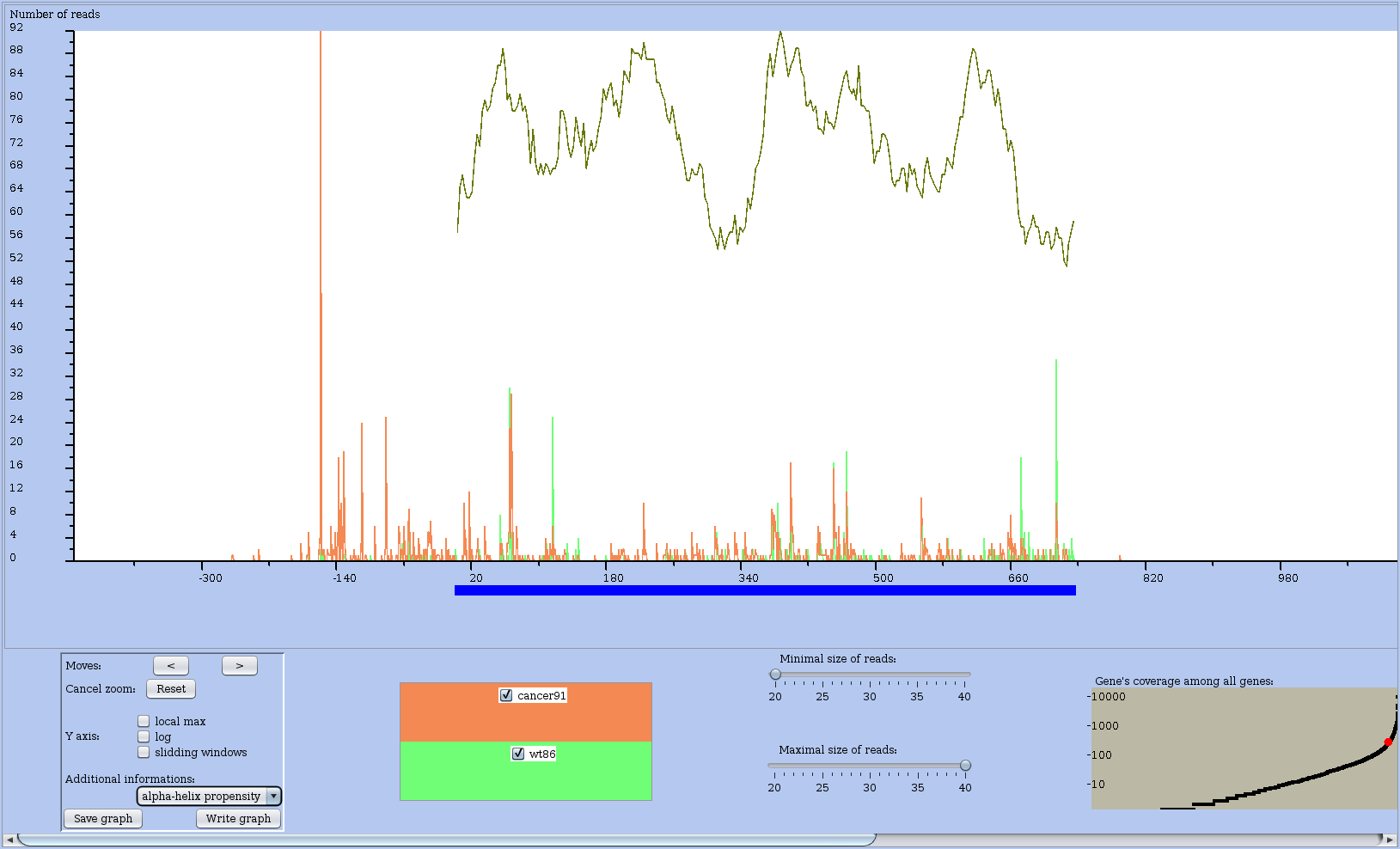
Figure 12: Coverage profile of a chosen RNA with the helix propensity curve shown on top of the RS profile in the CDS region.
Read size control panel (Figure 13)
With this panel, you can control the read sizes taken into account for the profile view. The two cursors (one for the minimum size and one for the maximum size) let you control the range of sizes used in the analysis. This is a specificity of very short read data and especially of RS data. Usually the fragments that are sequenced should be in a very restricted range on purpose. However, some mapped reads may be shorter / longer than expected and you can filter them out when needed.

Figure 13: Read size control panel in the center of a profile view of an RNA. Just set the minimum and maximum sizes of reads used for the profile view. The profile is updated on the fly to reflect the chosen size range.
Expression level comparative plot panel (Figure 14)
This plot indicates in terms of coverage how the selected RNA compare to all other RNA of this experiment. The red dot represents your RNA within the range of coverage which is drawn as a curve.
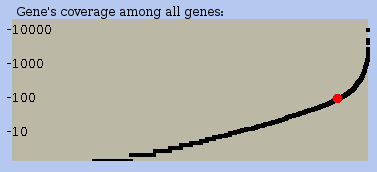
Figure 14: Lower right corner of a profile view of an RNA: a small plot indicates the expression level of the chosen RNA compared to all other RNA expressed in this dataset.
7 Comparative analyses
MA plot
In the Actions menu choose draw MA plot for this dataset against another dataset. It lets you choose to which dataset to compare and then a MA plot between these two experiments. The x-axis represents the mean RPKM between the two experiments. The y-axis represents ratio of RPKM values. RNAs are represented by blue points and one can click on a dot to obtain the profile view for that RNA. RRE will display the coverage of the selected RNA in another window. Indeed, clicking on a dot opens up the Details window for the corresponding RNA (see Figure 16), and shows its name, and some figures relative to the plot. It contains a button show coverage that allows you to get the profile view for this RNA.
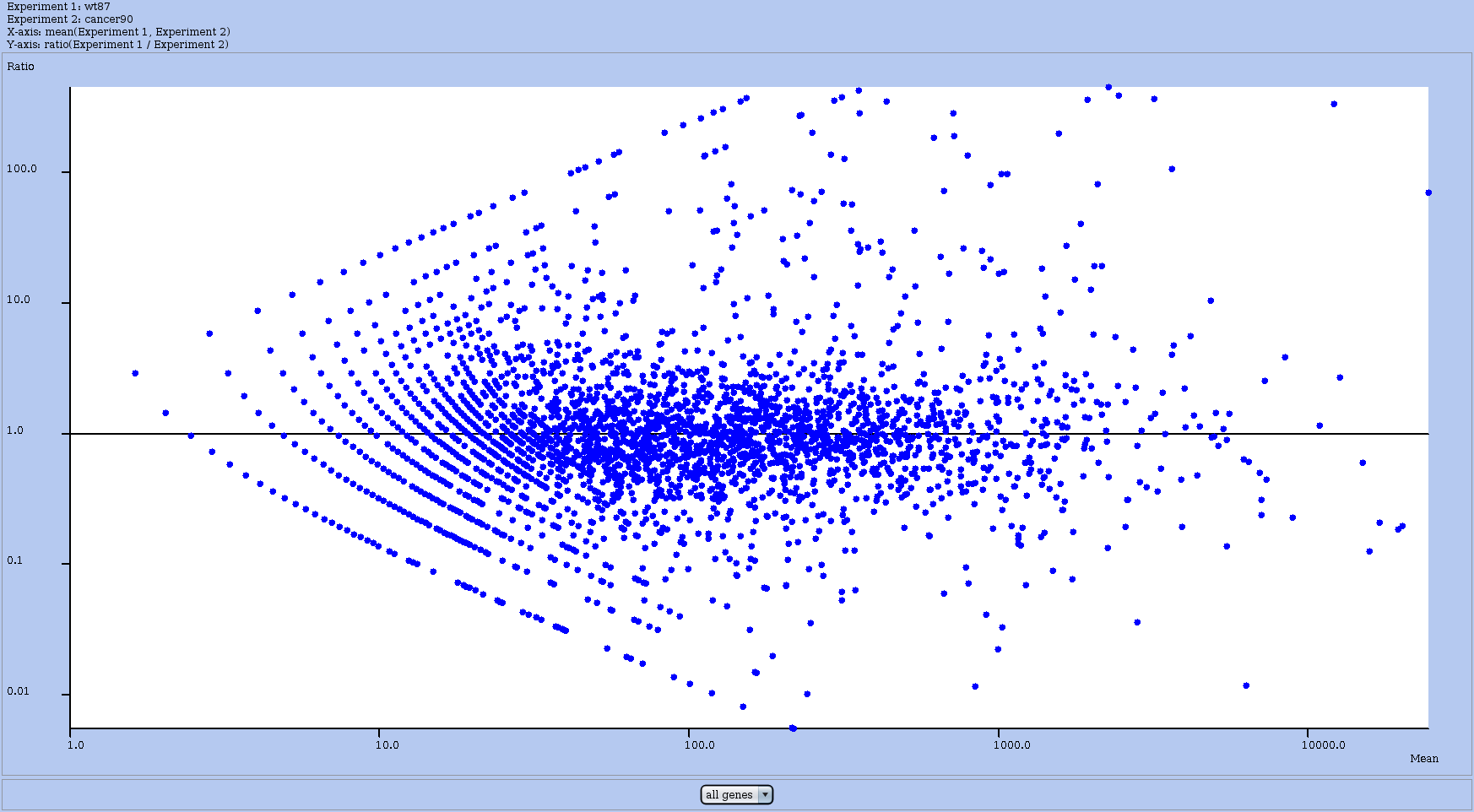
Figure 15: MA plot to compare globally two experiments. Each dot represents one RNA. Clicking on that dot opens up the Details window for it; from there you can ask for the profile view of that RNA.
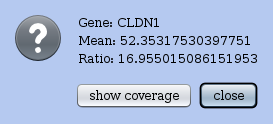
Figure 16: A Details window opens up when you click on a dot (i.e. an RNA) of a global plot (like the MA plot or the ORF-vs-URL plot).
Coding vs non-coding plot – or ORF vs UTR plot.
This analysis draw a plot that compares the coverages in the coding region (of the main ORF) vs in the untranslated regions (UTR) for all genes. Like in other global plots each dot is an RNA. See Figure 17.
Remarks:
- Reads that are not in the main ORF are in the UTR regions (either the 3' or the 5' UTR), and conversely.
- One should keep in mind that this is a global view: not all RNA are "equal" in the sense that the length of their main ORF and of their UTR vary across the set of RNAs.
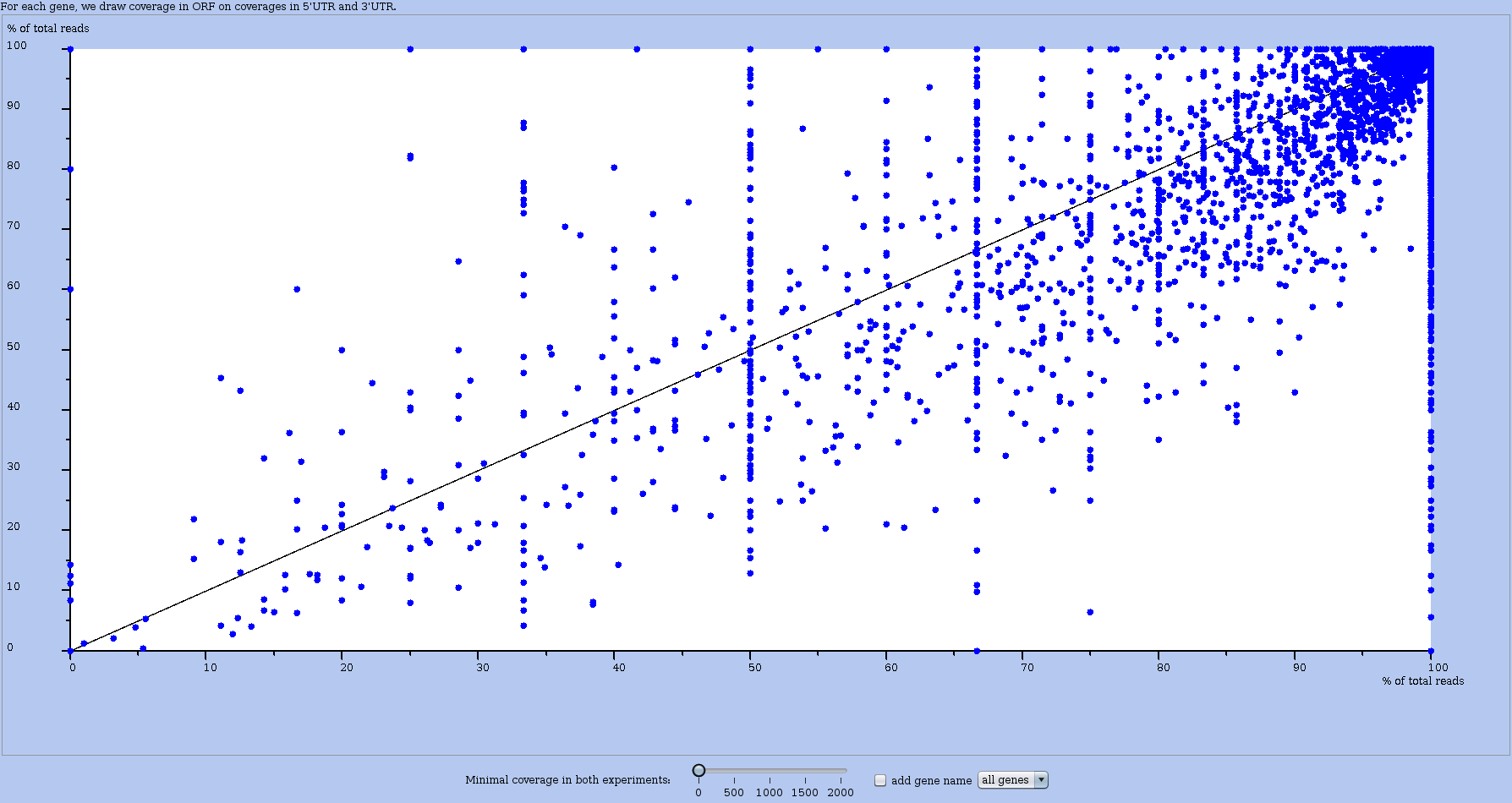
Figure 17: A global plot ORF vs UTR for comparing two datasets (or experiments). Each dot is an RNA. The axis represent the percentage of reads mapped in the main ORF region of that RNA in each experiment (one per axis). The dot of DYLN2 RNA shows a percentage of 56% in one experiment (X-axis), and of 33% in the other (Y-axis).
A graduated cursor below the plots controls the minimum coverage for an RNA to be on the plot. By default, all RNAs are considered. If you move the cursor to a non zero value, all RNAs whose coverage (in percents) in either experiment is below the threshold value, are removed from the plot. The effect of this control is shown in Figure 18.
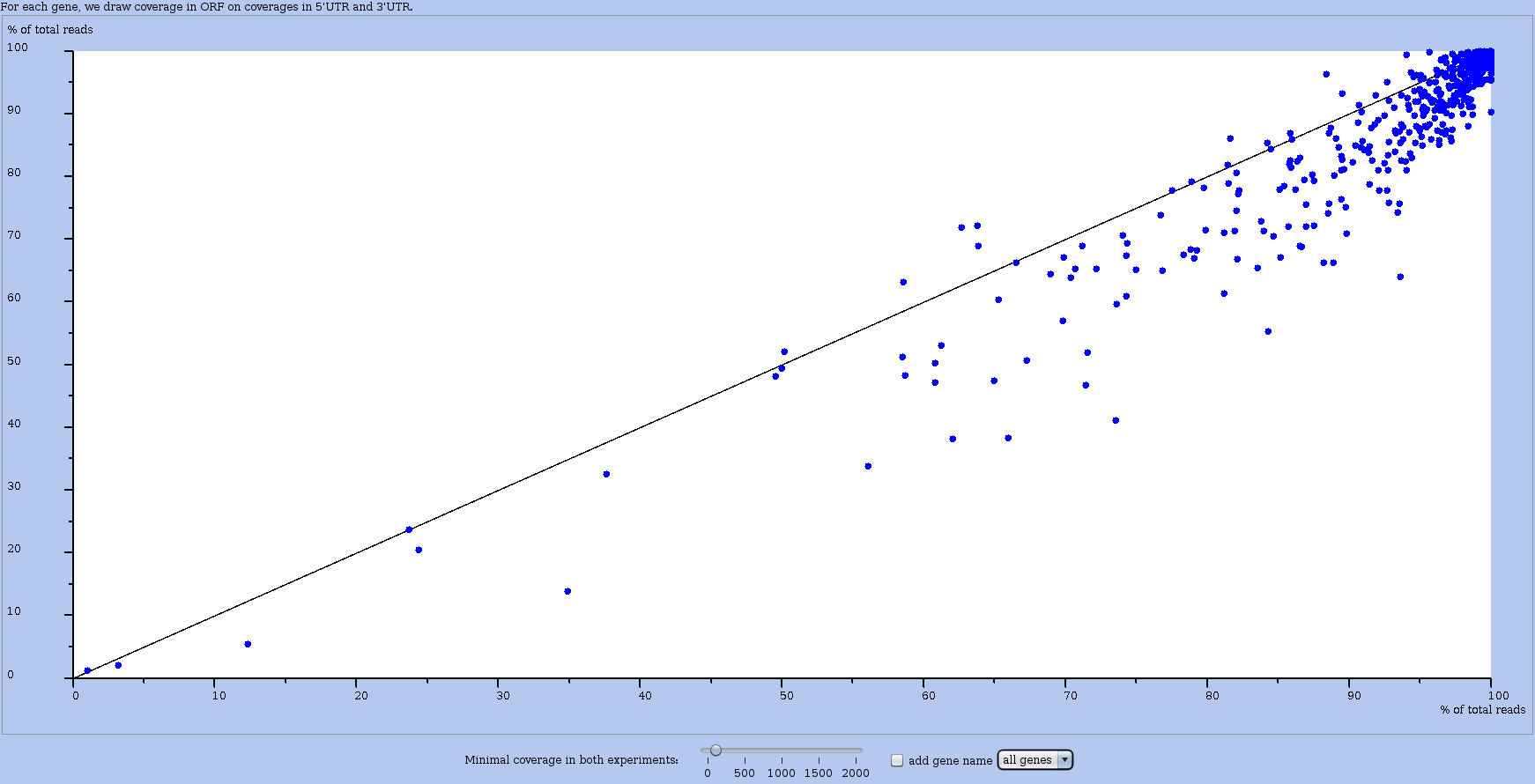
Figure 18: Another global plot ORF vs UTR for comparing two datasets (or experiments). Here, the minimum coverage cursor is set to a positive value. If you move the cursor, RRE updates the plot on-the-fly.
RPKM comparative plot
In the Actions menu, use the draw all genes vs another experiment to compute the RPKM plot. With it, you can compare the RPKM of all RNAs between two experiments. As in other global plots, an RNA is represented by a blue dot. An example is shown in Figure 19.
To have the axis in log-scale, toggle the log scale button. See the effect in Figure 20.
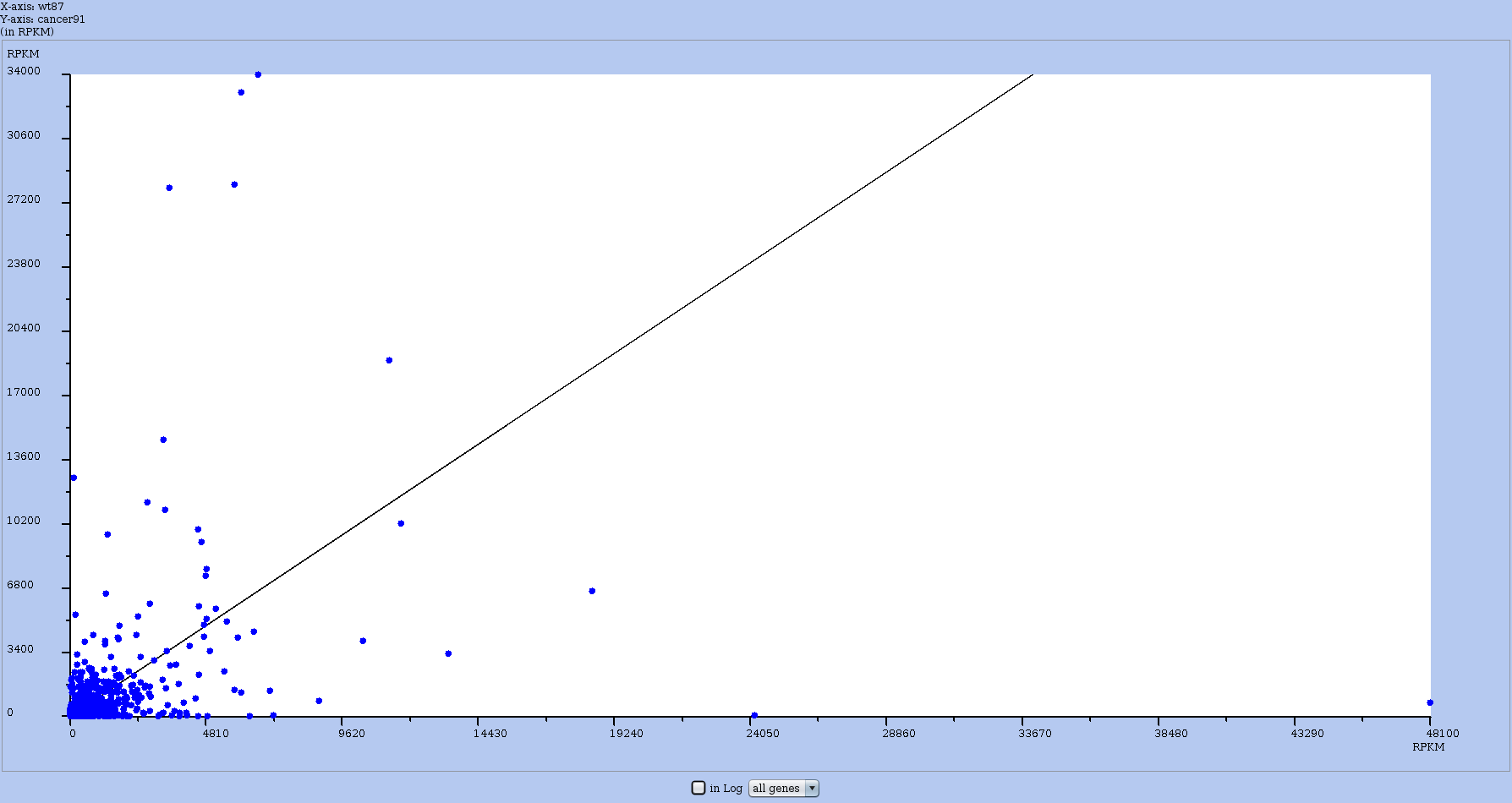
Figure 19: RPKM comparative plot that contrasts two datasets. Obtained from the Actions menu of one dataset, then one chooses the other dataset to compare to.
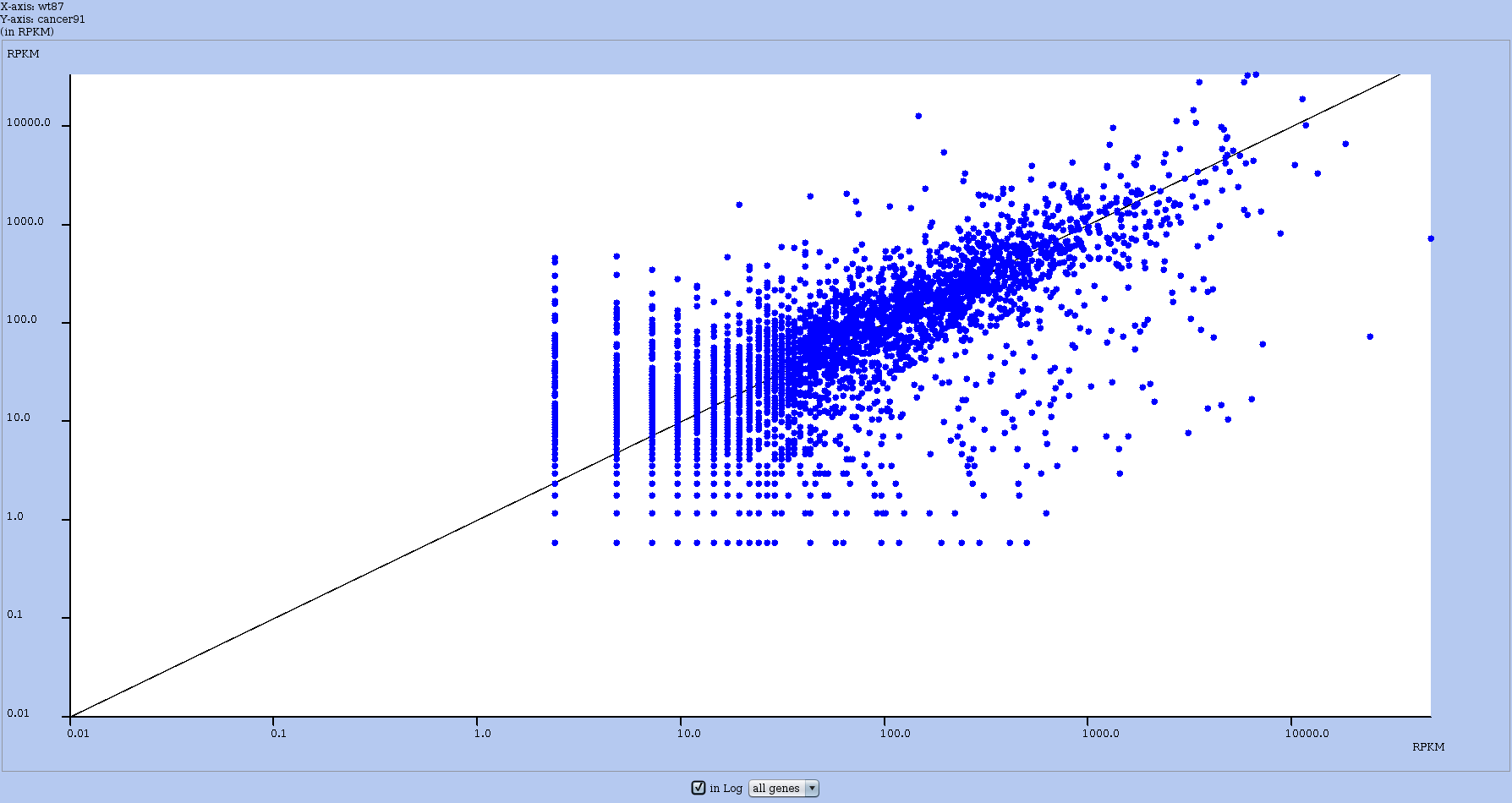
Figure 20: Same RPKM comparative plot as in with axis in log-scale.
Comparing user-defined regions of coverage
Analyses related to ORF and UTR are based on annotations (a GTF file used during the pre-processing of the read mapping results). Obviously, mRNA vary in overall length, in ORF length, and in UTR length. However, changes in translation may affect sub-regions of an RNA and alter this RNA profile in those sub-regions that do not exactly match annotated regions. Hence, RRE propose to contrast the coverage in two user-defined sub-regions.
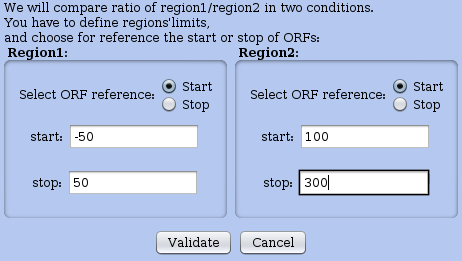
Figure 21: Window for defining specific regions in RNAs. The user can define regions that do not necessarily match annotated regions. The coverages of these regions will be contrasted in a global plot for the two chosen conditions (see plot in Figure 22).
For this, in the Actions menu choose the option compare regions of coverage. First choose the other dataset to contrast with. Then a second window appears; there you define your regions of interest by indicating the leftmost and rightmost nucleotidic positions for each sub-region. This occurs in a window shown in Figure 21, while the resulting plot appears in Figure 22. Note that the scale is logarithmic. In the footer of the plot window, you can adjust the overall coverage threshold thanks to a cursor, or use a previously saved selection to restrict the plot to a subset of RNAs of interest. The main diagonal line on the plot appears between to other diagonal that mark +5% and -5% of the coverage ratio.
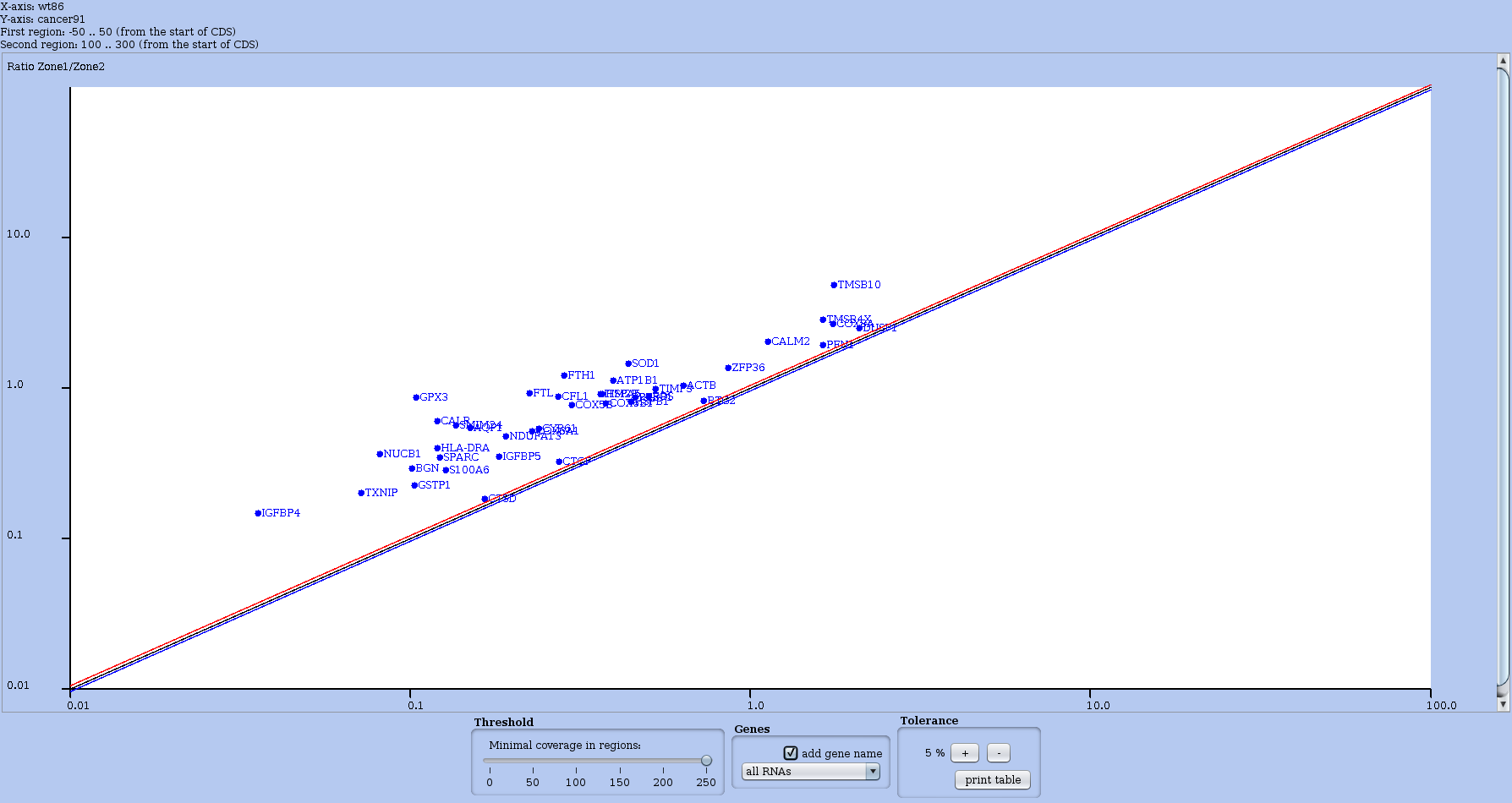
Figure 22: Global plot contrasting the coverage of user-defined regions (Figure 21). Note that the scale is logarithmic. Here, the overall coverage threshold has been set to 250 (thanks to the Threshold cursor in the footer of the plot window). Moreover, each RNA's name is written next to the corresponding dot on the plot.
Looking at the RS profile of RNA named FTL, in wild type (Figure 23) and in cancer condition (Figure 24), one observes that most important peaks in cancer are located in the 5' UTR up to nucleotide 50, while in the wild type condition high peaks are spread along the main ORF.
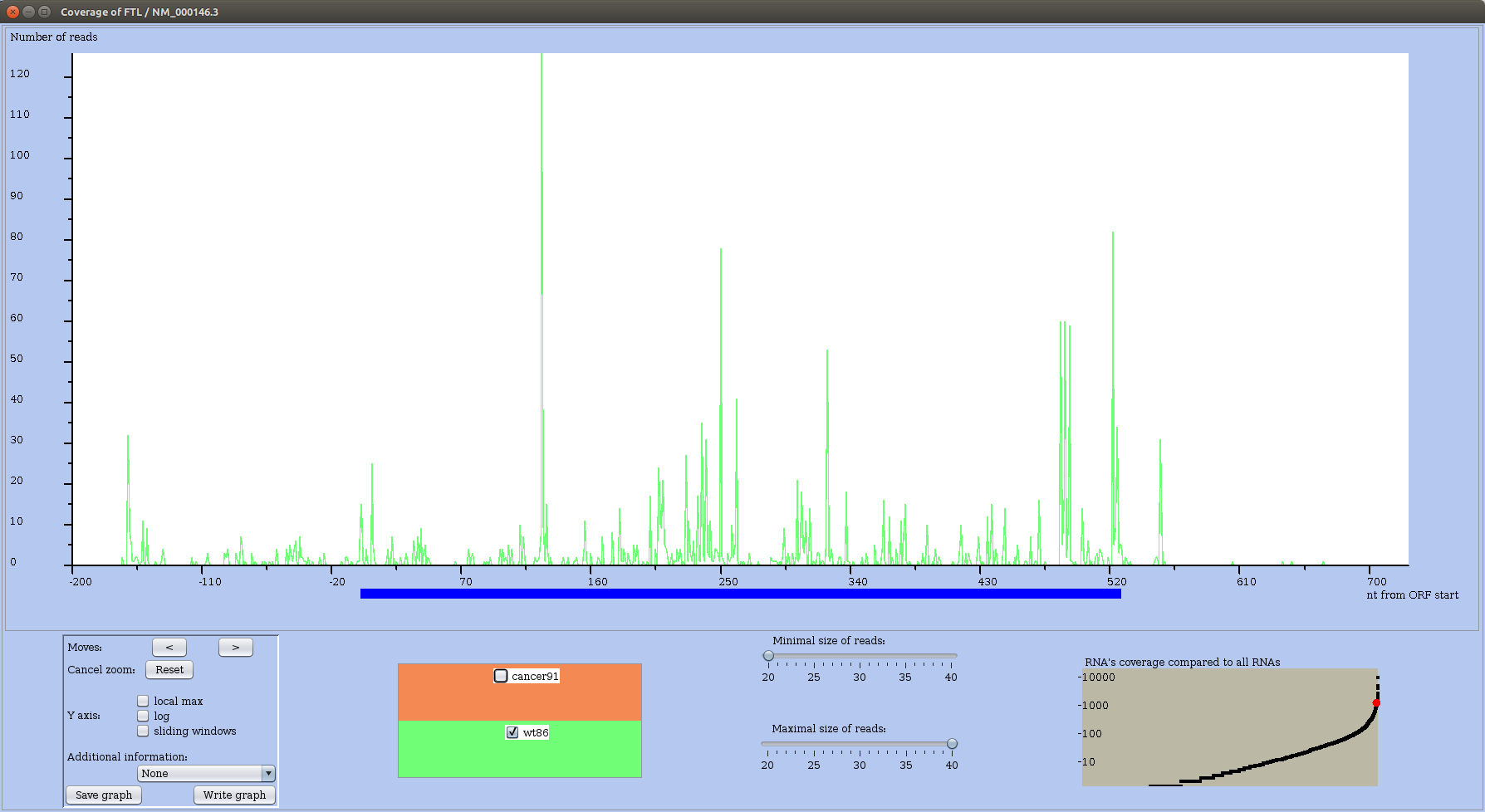
Figure 23: RS profile of FTL mRNA in the wild type condition.
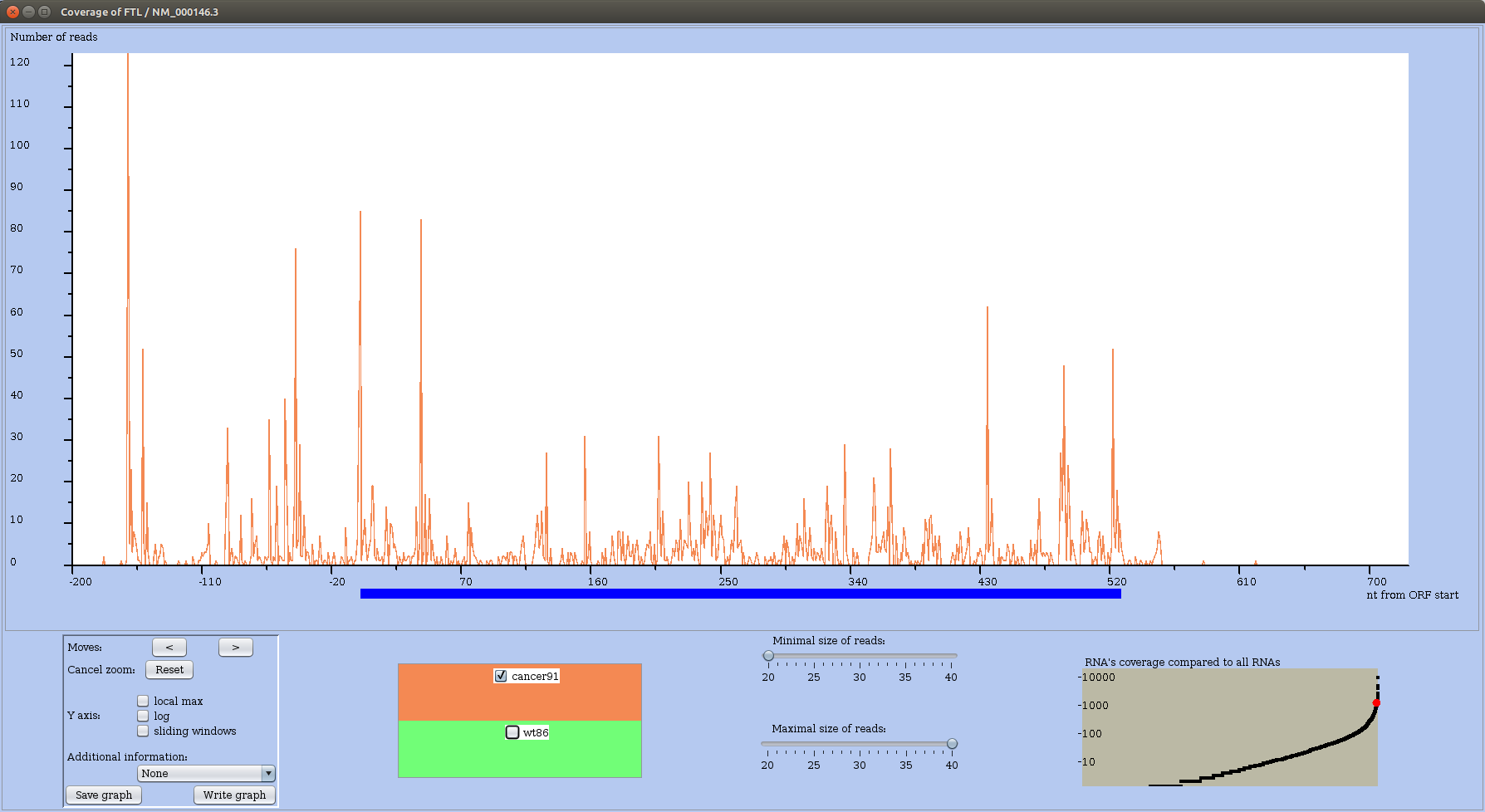
Figure 24: RS profile of FTL mRNA in the cancer condition.
Correlation plot between two datasets
For this, in the Actions menu (you may have to first drag the elevator down) choose the option compare correlation. RRE plots for each mRNA the correlation of coverages in both conditions (Y-axis) in function of the mean coverage (see Figure 25).
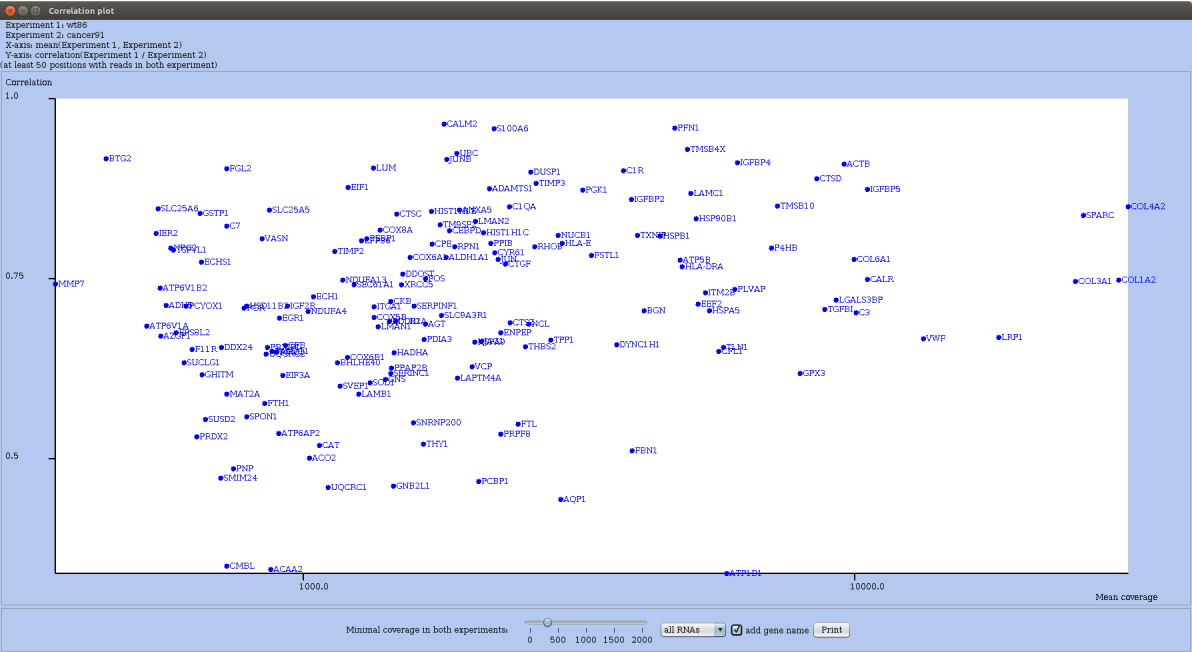
Figure 25: Correlation plot for a wild type and a cancer datasets. Global coverage of each mRNA is thresholded and gene names are shown next to each dot.
Comparing main phase in mRNAs across two datasets
For this, in the Actions menu (you may have to first drag the elevator down) choose the option compare main phase to another experiment. For each RNA, RRE draws the proportion of reads mapping in the main phase. The value for the reference experiment is on X-axis, while for the other experiment it is on Y-axis. In Figure 26, all RNAs in which at least 20% of codons are covered by reads are plotted. The color of the dot depends on the main phase (O in black, 1 in red and 2 in blue). Buttons allows you to select which "phase" is drawn on the plot. Unselecting phase 0, let you see the few RNAs whose main phase are 1 or 2 (see Figure 27).
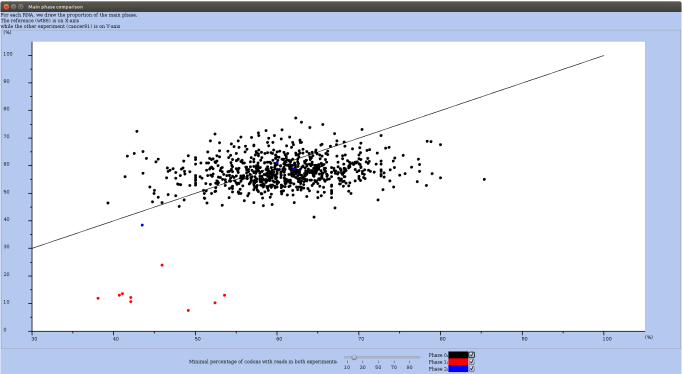
Figure 26: Window with the Main phase comparison plot.
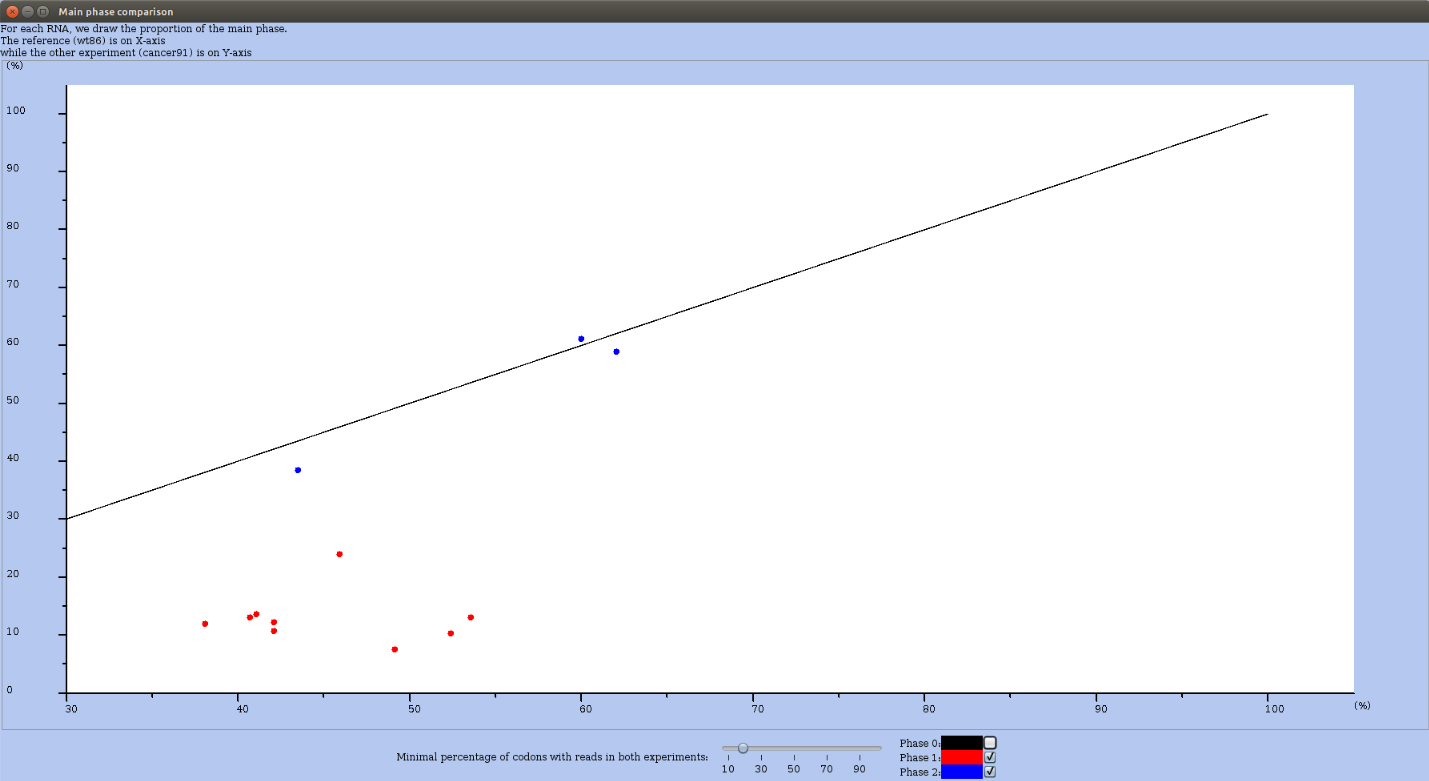
Figure 27: Main phase comparison plot (as in 26), where all RNAs whose main phase is 0 are hidden.
8 Mining and selecting interesting subsets of RNAs with queries
With the option Mine or select subsets of RNAs with queries from the main Analyses menu, you can mine interesting subsets of RNA with formal queries. By "interesting" we mean RNAs whose translation seems unusual with respect to annotation. One expects an RNA to have its main ORF translated; however, it occurs that alternative ORFs are translated instead and simultaneously to the main ORF. So, by setting one or more conditions on the RS coverage of RNAs, you form a query and let RRE search for all RNAs that satisfy those conditions.
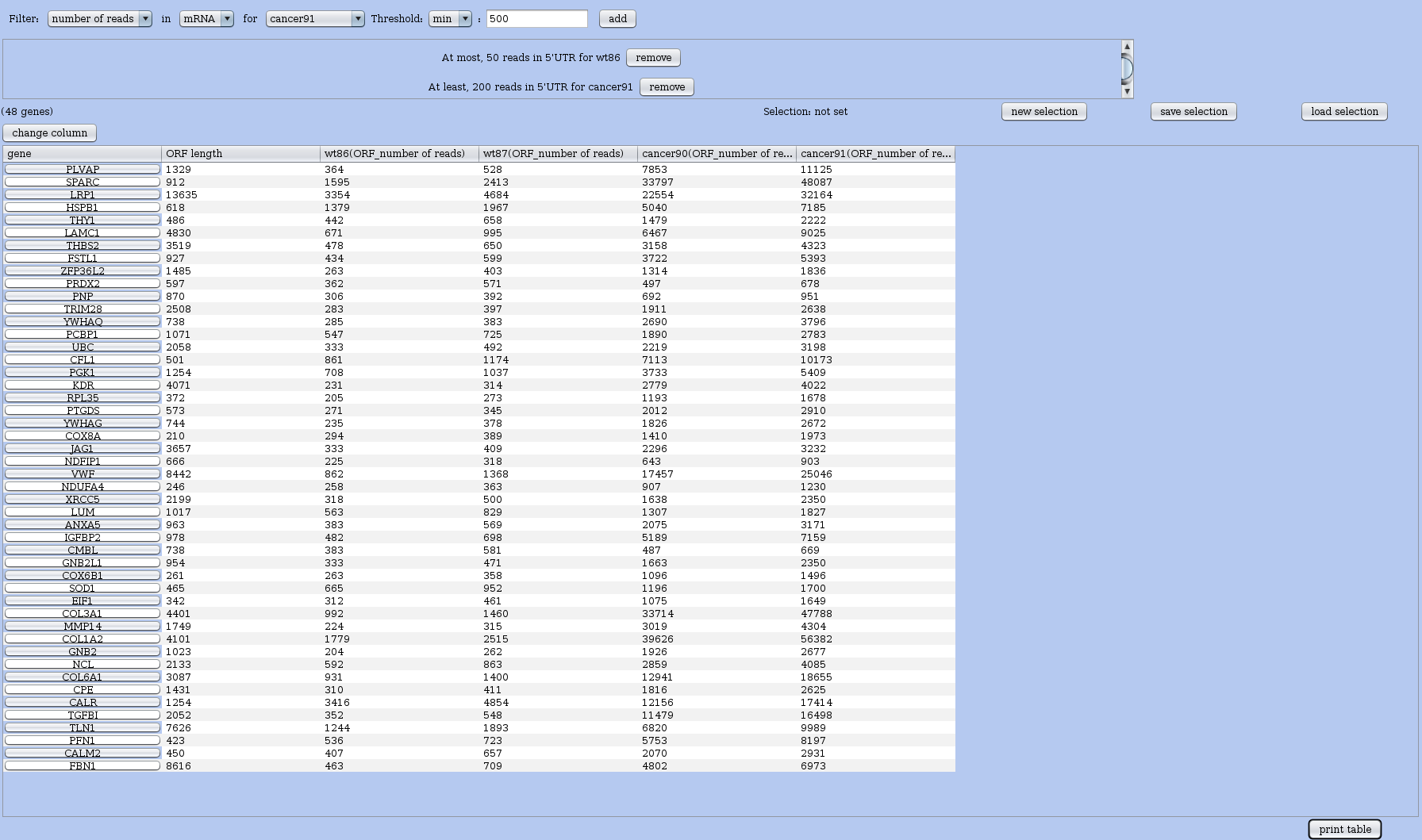
Figure 28: Window for mining subsets of RNAs. The query interface is on top, below it you see the interactive table of results. This table logically contains all RNAs at start (when there is no query). Once you have added conditions to the query, the results are recomputed and the table updated. You can save the table as an external file. You can also save and name this selection, and of course reload existing selection or reset the selection.
You can modify the information that appear in the table using the change columns button located just above the table – see Figure 29. The print table allows you save the content of the table in a file on the disk in tabular format for later use with any spreadsheet, statistical software, or the R language.

Figure 29: Window for changing the information columns in the result table for the current query.
The idea is to let RRE search for the RNAs that satisfies some conditions automatically, and to avoid doing that by visual inspection. This has two advantages:
- an algorithm computes exactly the subset of RNAs (while it is easy to miss some with human inspection)
- it is fast.
You can then save this subset as a selection, give it a name. It will be kept in memory and you can perform some of the above-mentioned analyses restricted to this subset by using the given name. The page footer with the selection menu is shown in Figure 30.

Figure 30: Footer of a plot window with the menu to choose either "All RNAs" or any user-defined, previously saved, selection. User-defined selections will appear in the rolling menu button.
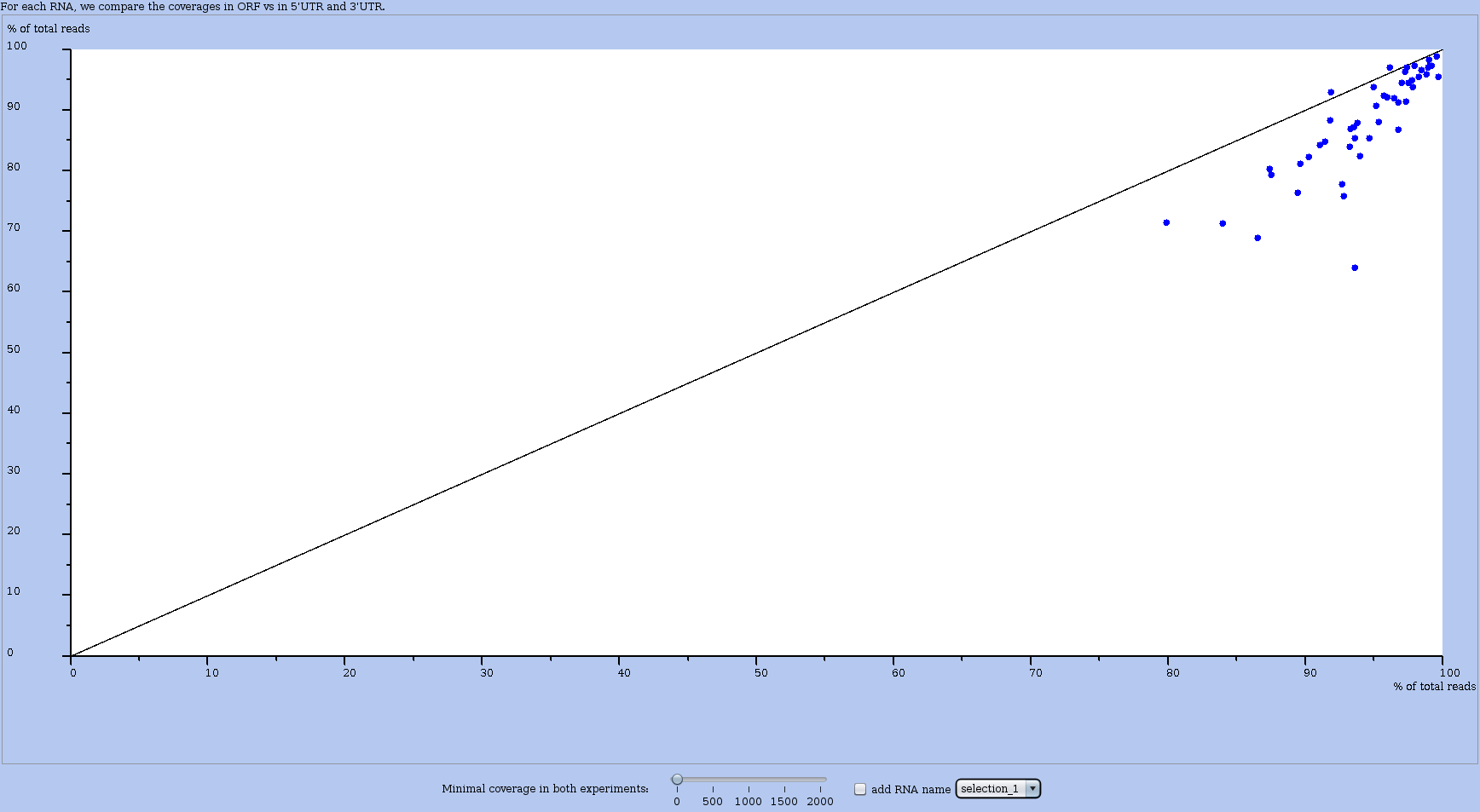
Figure 31: Window with an ORF vs UTR plot applied to a user-defined selection (here "selection 1"). Hence, not all RNAs are plotted, but only those belonging to the subset of RNAs corresponding to the chosen selection (see Figure 30). In other words, those RNAs that satisfy the conditions in the query (that defined the selection).
9 Contact, reference, and links.
Contact
For comments, support, feedback, requests, etc., please email translatome@lirmm.fr with your name, contact address.
Article / preprint
RNA-Ribo-Explorer: interactive mining and visualisation of Ribosome profiling data
D. Paulet, A. David, E. Rivals
HAL reference lirmm-01998415v1, 9 p., Jan. 2019.
Links
Complementary tools and pipelines We have developed a tool to assess codon usage bias using RS data. Please have a look at: RSCURS webpage.
If you search for a pipeline to process RS data on a reference genome or reference transcriptome, please contact us at translatome@lirmm.fr
Support
We acknowledge the support of the Institut de Biologie Computationnelle (ANR-11-BINF-0002), and of the GEM project financed through Labex NUMEV. We thanks ATGC bioinformatics platform for hosting the software page, as well as the Institut Français de Bioinformatique for general support to the platform.
![]()
Figure 32: Current support for hosting and maintaining RRE is from the ATGC bioinformatics platform.
![]()
Figure 33: Funding support from Institut de Biologie Computationnelle (IBC).
![]()
Figure 34: Funding support from GEM Flagship project from Labex NUMEV.
![]()
Figure 35: Funding support from the French National Cancer Institute (INCA).