FastME 2.0
FastME is a software package for the fast and accurate inference of phylogenetic trees from distance matrices. It implements algorithms based on the Balanced Minimum Evolution (BME) principle, a distance-based criterion closely related to the Neighbor Joining (NJ) method. The goal of the BME framework is to identify the phylogenetic tree that minimizes the total estimated evolutionary distance among taxa.
Compared to classical Neighbor Joining approaches, FastME improves tree accuracy through efficient topological optimization procedures. After constructing an initial tree, the program refines the topology using rearrangement operations such as Nearest Neighbor Interchange (NNI) and Subtree Pruning and Regrafting (SPR). These optimization steps allow FastME to explore alternative tree topologies and identify trees with shorter total branch lengths while maintaining a computational cost comparable to NJ.
FastME supports multiple types of input data, including DNA sequences, protein sequences, or precomputed distance matrices, and provides several methods for evolutionary distance estimation. The software also includes features such as bootstrap analysis for branch support, parallel computation, and several tree refinement strategies.
Thanks to its combination of speed, scalability, and accuracy, FastME is widely used for phylogenetic reconstruction, particularly for large datasets where likelihood-based approaches may be computationally demanding.
Reference: Lefort V., Desper R., Gascuel O. (2015). FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Molecular Biology and Evolution, 32(10), 2798–2800.
FastME 2.0 online execution

Other tools

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
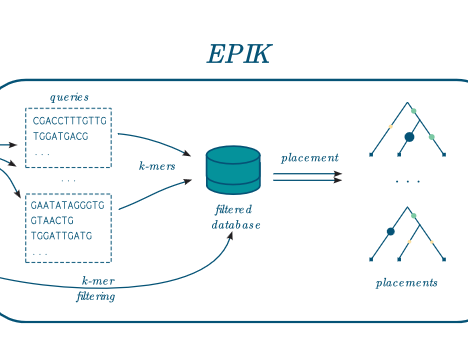
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…