DExTER
Overview
DExTER (Domain Exploration To Explain gene Regulation) is a bioinformatics tool designed to automatically identify genomic regions whose nucleotide composition correlates with gene expression levels.
Unlike traditional approaches focusing on short transcription factor binding sites (6-12 bp), DExTER detects Long Regulatory Elements (LREs) that can span tens to hundreds of nucleotides.
This makes it possible to explore a poorly characterized regulatory mechanism: gene regulation driven by the global composition of genomic regions.
The method has shown strong predictive power, explaining a large fraction of gene expression variability, reaching more than 70% prediction accuracy in Plasmodium falciparum.
Method principle
DExTER combines DNA sequences and gene expression data to identify pairs:
(k-mer motif, gene region)
whose frequency is correlated with expression level.
Main steps
- Segmentation of regions around genes
- Iterative search for k-mers correlated with expression
- Informative variable selection (LASSO / machine learning)
- Construction of a predictive gene expression model
The final model allows:
- identification of candidate regulatory regions
- explanation of expression variability
- prediction of expression of new genes
Biological applications
DExTER enables the study of regulatory mechanisms not detectable with classical motif discovery methods:
- Genomes with few transcription factors
- Post-transcriptional regulation
- Cell-cycle dependent regulation
- Epigenetic regulation linked to DNA composition
Key observations from the study:
- Highly dynamic regulation in Apicomplexa
- More stable regulation in multicellular organisms
- Distinct roles of upstream (transcriptional) vs downstream (post-transcriptional) regions
Input data
- Nucleotide sequences aligned per gene (e.g. ±2 kb around TSS or start codon)
- Gene expression matrix (RNA-seq or microarray)
Output data
- List of candidate regulatory elements (cLREs)
- Enriched motifs and associated regions
- Variable importance
- Predictive expression model
- Correlation scores between sequence and expression
You can explore an example of the results generated by DExTER here:
https://api.atgc-montpellier.fr/results/71cb194e-4dc4-4b4d-aca7-f153659cd340/
Typical use cases
- Study of non-canonical gene regulation
- Comparative genomics
- Transcriptome interpretation
- Atypical genomes (parasites, plants, protists)
Associated publication
Menichelli C. et al., 2021
Identification of long regulatory elements in the genome of Plasmodium falciparum and other eukaryotes
PLOS Computational Biology
https://doi.org/10.1371/journal.pcbi.1008909
Source code
https://gite.lirmm.fr/menichelli/DExTER
DExTER online execution

Other tools
PhyML 3.0
Overview: new algorithms, methods and utilities PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient…

AQUAPONY
AquaPony: interactive visualization of phylogeographic scenarios AquaPony is a web application designed to explore and interpret evolutionary scenarios on annotated phylogenetic trees (for example, ancestral geographic states). It was built to make uncertainty in ancestral reconstructions easier to understand and communicate. Why AquaPony? In phylogeography, several scenarios can be nearly as plausible as the best…
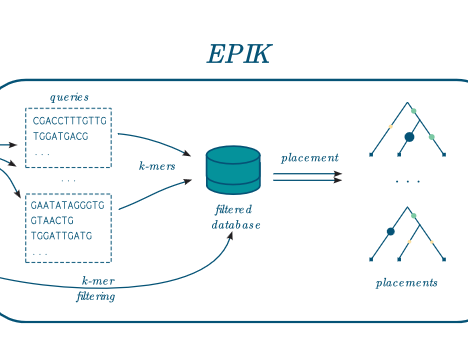
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…