AQUAPONY
AquaPony: interactive visualization of phylogeographic scenarios
AquaPony is a web application designed to explore and interpret evolutionary scenarios on annotated phylogenetic trees (for example, ancestral geographic states). It was built to make uncertainty in ancestral reconstructions easier to understand and communicate.
Why AquaPony?
In phylogeography, several scenarios can be nearly as plausible as the best one. AquaPony helps users go beyond a single “best” reconstruction by interactively examining alternative scenarios under user-defined uncertainty thresholds.
Key features
- Interactive visualization of the full tree and selected subtrees.
- Explicit display of uncertainty in ancestral state assignments.
- Exploration of alternative phylogeographic scenarios along branches.
- Instant visual updates, including for large trees.
- Export-ready figures for reports and publications.
Use cases
AquaPony is relevant to infectious disease research, ecology, agronomy, and more broadly to any study involving spatiotemporal evolutionary inference from annotated phylogenies.
Access the tool
Open AquaPony here: https://old.atgc-montpellier.fr/aquapony/aquapony.html
Scientific reference
Cazaux B., Castel G., Rivals E. (2019). AQUAPONY: visualization and interpretation of phylogeographic information on phylogenetic trees. Bioinformatics, 35(17), 3163–3165. DOI: 10.1093/bioinformatics/btz011

Other tools

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
LoRDEC: hybrid correction of long…
Overview In a nutshell, LoRDEC is a program for error correcting long sequencing reads using short reads. It implements a hybrid correction approach. It uses little memory and is very efficient. Most importantly it scales up to process very large data sets. It can be applied to long reads obtained with either Pacific Biosciences SMRT…
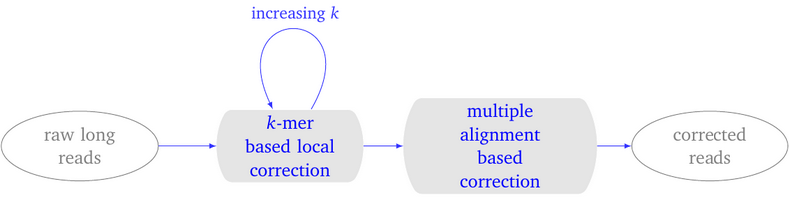
LoRMA: a self correction program…
Overview LoRMA is an error correction program for long reads, which are sequences obtained using the third generation of sequencing technologies (3GS), either with Oxford Nanopore technology or with Pacific Biosciences technology. LoRMA is a so-called self-correction software, as opposed to e.g. LoRDEC that is a hybrid error correction tool. This means that LoRMA uses…