RSCU_RS: Measuring the bias in codon usage from ribosomal activity
Overview
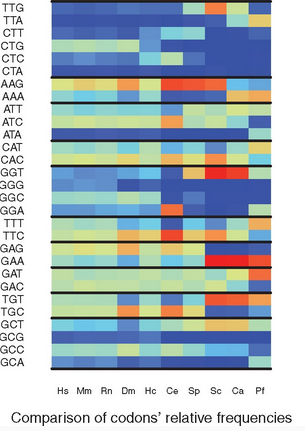
Overview: In the protein coding sequences of a species, the 61 possible codons of the genetic code are not equally distributed. This observation is referred to as the Codon Usage Bias (CUB) of a species. Several measures have been proposed to quantify the CUB using the frequencies of codons in all RNA coding sequences (or at least a representative subset of these). This yields a static analysis since sequences change slowly over time. But in living cells the translation varies over time, since the ribosome, the molecular machine that performs translation, has a role in the selection of which RNA sequences are indeed translated. The precise location of ribosomes translating RNA can be monitor by Ribo-seq (aka ribosome profiling), a high throughput sequencing assay that captures the portions of RNA located inside the ribosomes. Thus, the sequencing reads produced by Ribo-seq in a given condition give us access to which codons ribosomes are translating.
We proposed to measure the codon usage bias in a transcriptome wide manner using Ribo-seq sequencing data. This delivers a dynamic and precise estimation of Codon Usage Bias, since it integrates the location of ribosomes during translation. The codon usage bias can be computed classicaly for a given species, for a subset of genes, but also for any given condition for which Ribo-seq data is available. The CUB can thus be compared across species, across subsets of genes, or across conditions.
For this, we develop a stand-alone software, called RSCU_RS, and report experiments in estimating and comparing CUB across species in a journal article.
Reference
Journal article
Ribo-seq enlightens Codon Usage Bias
D. Paulet, A. David, E. Rivals
DNA Research, dsw062. https://doi.org/10.1093/dnares/dsw062, 2017.

Documentation and ressources
A documentation about the software RSCU_RS is available at
https://www.lirmm.fr/~rivals/rscu/
Funding
Fondation pour la Recherche Médicale, grant DBI20131228574.
ANR grant (ANR-11-BINF-0002), Institut de Biologie Computationnelle (IBC).
Other tools

MYST : Manage Your Scientific…
What is MYST? MYST is the orchestration platform behind ATGC online bioinformatics services. It provides a unified web interface and a public REST API to submit analyses, monitor jobs, and retrieve results across a growing catalog of phylogenetic and sequence-analysis tools. MYST is a modernized redesign of WAVES, an older tool previously developped by ATGC…
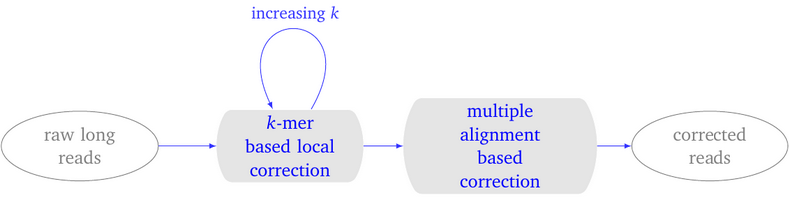
LoRMA: a self correction program…
Overview LoRMA is an error correction program for long reads, which are sequences obtained using the third generation of sequencing technologies (3GS), either with Oxford Nanopore technology or with Pacific Biosciences technology. LoRMA is a so-called self-correction software, as opposed to e.g. LoRDEC that is a hybrid error correction tool. This means that LoRMA uses…

WAVES
Summary WAVES is a web application dedicated to bioinformatic tool integration. It provides an efficient way to implement a service for any bioinformatic software. Such services are automatically made available in three ways: web pages, web forms to include in remote websites, and a RESTful web services API to access remotely from applications. In order…