EPIK: Precise and scalable evolutionary placement with informative k-mers
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree.
It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS remains valid.
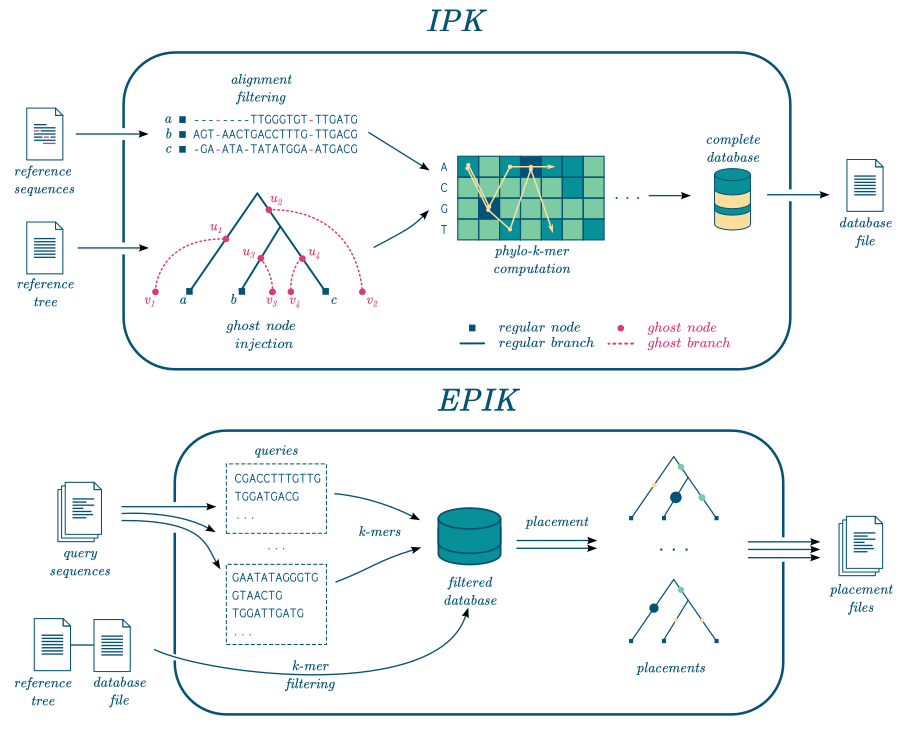
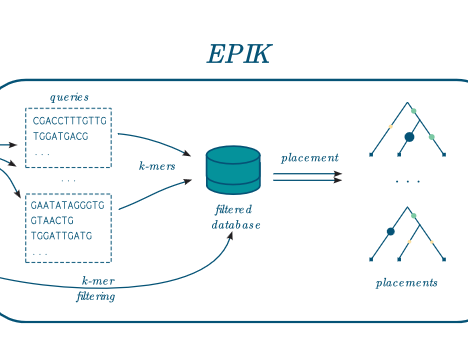
EPIK takes as input a file containing database of phylo-k-mers built with IPK and a set of reads to place on the related phylogeny. It works for nucleotidic and amino-acid query sequences.
EPIK can filter the database to load only the most informative phylo-k-mers, which reduces the memory usage.
EPIK can also run in parallel.
Keywords
Phylogenetic placement, metabarcoding, taxonomic identification, NGS, software
Other tools

FastME 2.0
FastME is a software package for the fast and accurate inference of phylogenetic trees from distance matrices. It implements algorithms based on the Balanced Minimum Evolution (BME) principle, a distance-based criterion closely related to the Neighbor Joining (NJ) method. The goal of the BME framework is to identify the phylogenetic tree that minimizes the total…

DExTER
Overview DExTER (Domain Exploration To Explain gene Regulation) is a bioinformatics tool designed to automatically identify genomic regions whose nucleotide composition correlates with gene expression levels. Unlike traditional approaches focusing on short transcription factor binding sites (6-12 bp), DExTER detects Long Regulatory Elements (LREs) that can span tens to hundreds of nucleotides. This makes it…
LoRDEC: hybrid correction of long…
Overview In a nutshell, LoRDEC is a program for error correcting long sequencing reads using short reads. It implements a hybrid correction approach. It uses little memory and is very efficient. Most importantly it scales up to process very large data sets. It can be applied to long reads obtained with either Pacific Biosciences SMRT…