PhyML 3.0
Overview: new algorithms, methods and utilities
PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient options to search the space of phylogenetic tree topologies.
Installation
To install PhyML, download the code from https://github.com/stephaneguindon/phyml/. After unpacking the archive, go into the phyml/ folder and type the following command:
sh ./autogen.sh;If you are using a Mac computer or running a Unix-like operating system, you will need to install the packages autoconf automake and pkg-config. On a Mac, the following command should set you up (provided Homebrew is installed on your Mac…): brew install pkg-config autoconf automake;
Next, to install any program that is part of the PhyML package, type the following commands:
./configure --enable-phyml;
make;To compile a Windows executable, install MinGW and run:
./configure --enable-win --enable-phyml;
make;To install the MPI version of PhyML, type the following commands:
autoreconf -i;
./configure --enable-phyml-mpi;
make;PhyML 3.0 online execution
Other tools

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
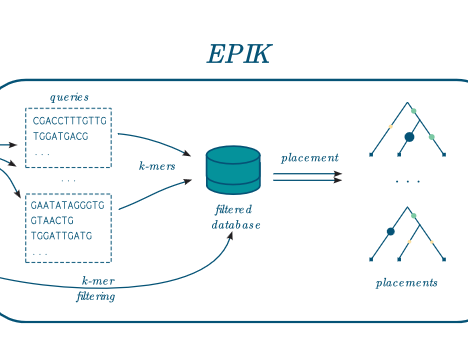
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…
LoRDEC: hybrid correction of long…
Overview In a nutshell, LoRDEC is a program for error correcting long sequencing reads using short reads. It implements a hybrid correction approach. It uses little memory and is very efficient. Most importantly it scales up to process very large data sets. It can be applied to long reads obtained with either Pacific Biosciences SMRT…