PhyML 3.0
Overview: new algorithms, methods and utilities
PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient options to search the space of phylogenetic tree topologies.
Installation
To install PhyML, download the code from https://github.com/stephaneguindon/phyml/. After unpacking the archive, go into the phyml/ folder and type the following command:
sh ./autogen.sh;If you are using a Mac computer or running a Unix-like operating system, you will need to install the packages autoconf automake and pkg-config. On a Mac, the following command should set you up (provided Homebrew is installed on your Mac…): brew install pkg-config autoconf automake;
Next, to install any program that is part of the PhyML package, type the following commands:
./configure --enable-phyml;
make;To compile a Windows executable, install MinGW and run:
./configure --enable-win --enable-phyml;
make;To install the MPI version of PhyML, type the following commands:
autoreconf -i;
./configure --enable-phyml-mpi;
make;PhyML 3.0 online execution
Other tools

AQUAPONY
AquaPony: interactive visualization of phylogeographic scenarios AquaPony is a web application designed to explore and interpret evolutionary scenarios on annotated phylogenetic trees (for example, ancestral geographic states). It was built to make uncertainty in ancestral reconstructions easier to understand and communicate. Why AquaPony? In phylogeography, several scenarios can be nearly as plausible as the best…
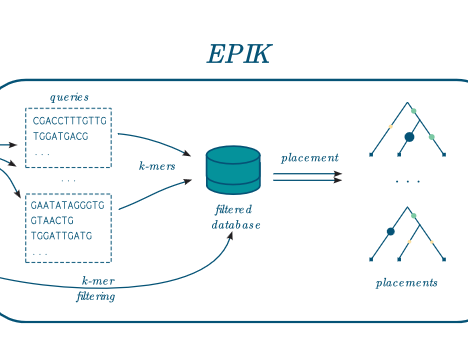
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…

FastME 2.0
FastME is a software package for the fast and accurate inference of phylogenetic trees from distance matrices. It implements algorithms based on the Balanced Minimum Evolution (BME) principle, a distance-based criterion closely related to the Neighbor Joining (NJ) method. The goal of the BME framework is to identify the phylogenetic tree that minimizes the total…