EPIK: Precise and scalable evolutionary placement with informative k-mers
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree.
It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS remains valid.
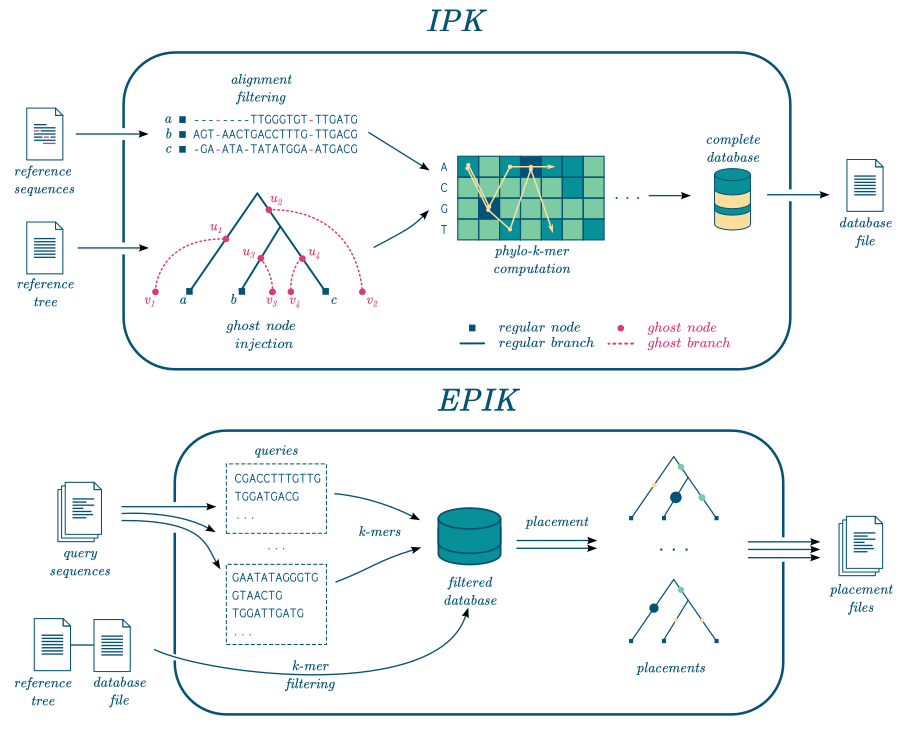
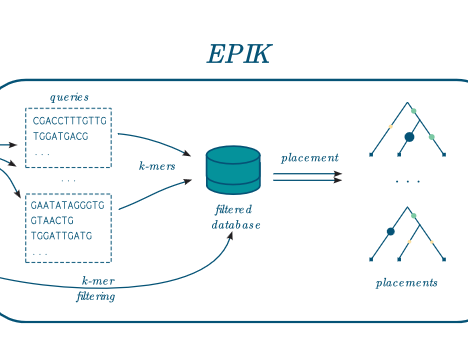
EPIK takes as input a file containing database of phylo-k-mers built with IPK and a set of reads to place on the related phylogeny. It works for nucleotidic and amino-acid query sequences.
EPIK can filter the database to load only the most informative phylo-k-mers, which reduces the memory usage.
EPIK can also run in parallel.
Keywords
Phylogenetic placement, metabarcoding, taxonomic identification, NGS, software
Other tools

TFscope
Characterizing the binding preferences of transcription factors (TFs) in different cell types and conditions is key to understand how they orchestrate gene expression. TFscope is a machine learning approach that identifies sequence features explaining the binding differences observed between two ChIP-seq experiments targeting either the same TF in two conditions or two TFs with similar…

MYST : Manage Your Scientific…
What is MYST? MYST is the orchestration platform behind ATGC online bioinformatics services. It provides a unified web interface and a public REST API to submit analyses, monitor jobs, and retrieve results across a growing catalog of phylogenetic and sequence-analysis tools. MYST is a modernized redesign of WAVES, an older tool previously developped by ATGC…

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…