PEWO: a collection of workflows to benchmark phylogenetic placement
Introduction and context
In the Bioinformatics team of the LIRMM (CNRS & Univ. Montpellier), we develop a series of tools for metagenomics / metabarcoding analysis. Our tools exploit phylo-k-mers (which are k-mers combined with phylogenetic information) computed for an input set of reference sequences and their phylogeny. The phylo-k-mers are computed and indexed with IPK, which stores them in files. Then, using such a phylo-k-mer index (or database):
- EPIK can perform phylogenetic placement of an input set of metabarcoding reads
- SHERPAS identifies reads that are recombinant between different virus strains.
PEWO is a tool to execute, evaluate and compare virtually any tool that does phylogenetic placement of sequencing reads (currently these include Pplacer, EPA, EPA-ng, APPLES, AppSpam, RAPPAS and EPIK).
PEWO: overview

PEWO stands for Phylogenetic placement Evaluation WOrkflows.
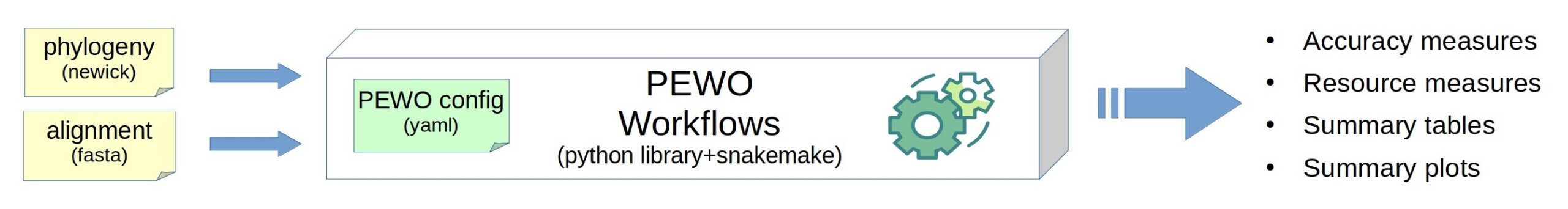
PEWO is a framework to run evaluation and comparison of any tool that performs phylogenetic placement of metagenomic reads. PEWO allows automatic evaluation of precision, of running time and memory usage for several tools on benchmark datasets. It runs the software, collect the results, and prepare graphics for ready-to-use figures. It evaluates all tools in a standard and carefully design procedure: that way you can run a fair comparison, but you can also explore which parameter setting best fits your use-case.
PEWO is freely accessible at https://github.com/phylo42/PEWO and comes with a Wiki, a tutorial, a comprehensive documentation and benchmark datasets.
In 2026, it includes 3 workflows: Pruning-based Accuracy evaluation (PAC), Likelihood-based Accuracy evaluation (LAC), and Resources evaluation (RES).
PEWO is a collaborative effort: test it and make it yours!
Technical overview
PEWO is a framework developped with Snakemake, Python, and Conda (for the management of environments). It already incoporates 7 published, standard tools for phylogenetic placement (see list above). PEWO is flexible and extensible: there is a ligthweight procedure to incorporate a new placement tool. PEWO was published in 2020 and adopted by the community that has already extended it.
License: MIT license.
Publication

Funding
France Génomique [ANR-10-INBS-0009], MNERT fellowship

Other tools
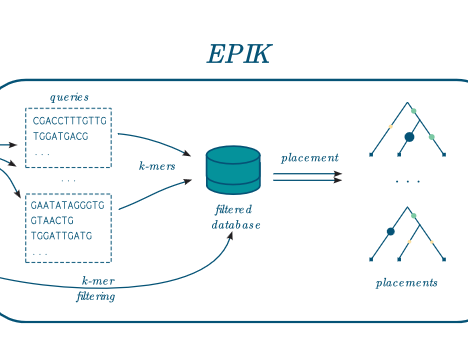
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…

WAVES
Summary WAVES is a web application dedicated to bioinformatic tool integration. It provides an efficient way to implement a service for any bioinformatic software. Such services are automatically made available in three ways: web pages, web forms to include in remote websites, and a RESTful web services API to access remotely from applications. In order…
LoRDEC: hybrid correction of long…
Overview In a nutshell, LoRDEC is a program for error correcting long sequencing reads using short reads. It implements a hybrid correction approach. It uses little memory and is very efficient. Most importantly it scales up to process very large data sets. It can be applied to long reads obtained with either Pacific Biosciences SMRT…