WAVES
Summary
WAVES is a web application dedicated to bioinformatic tool integration. It provides an efficient way to implement a service for any bioinformatic software. Such services are automatically made available in three ways: web pages, web forms to include in remote websites, and a RESTful web services API to access remotely from applications. In order to fulfill the service’s computational needs, WAVES can perform computation on various resources and environments, such as Galaxy instances.
Availability and implementation
WAVES was developed with Django, a Python-based web framework. It was designed as a reusable web application. It is fully portable, as only a Python installation is required to run Django. It is licensed under GNU General Public License version 3.
WAVES components
WAVES-core
This is the main WAVES component.
- GitHub repository: https://github.com/lirmm/waves-core
- Documentation: http://waves-core.readthedocs.io
- Installation guide: http://waves-core.readthedocs.io/en/latest/installation.html
- Administration guide: http://waves-core.readthedocs.io/en/latest/user_doc/user_guide.html
- Tutorial to create a ‘Hello world’ service: http://waves-core.readthedocs.io/en/latest/user_doc/howto/howto.html
- Developer guide: http://waves-core.readthedocs.io/en/latest/dev_doc/dev_doc.html
- Python code examples to interact with WAVES API: http://waves-core.readthedocs.io/en/latest/dev_doc/sample_code.html
WAVES-Galaxy
This is a WAVES adapter dedicated to Galaxy. Using the BioBlend python library, this adapter enables Galaxy services import into WAVES-core. WAVES-Galaxy automatically recognizes the tools available in a Galaxy instance.
- GitHub repository: https://github.com/lirmm/waves-galaxy
- Documentation: http://waves-galaxy-adaptors.readthedocs.io
WAVES-demo
This is a custom WAVES instance for demo purpose. It was created to show what could be done with Django functionalities to custom a WAVES installation.
- GitHub repository: https://github.com/lirmm/waves-demo
- Documentation: http://waves-demo.readthedocs.io
- Demo: http://waves.demo.atgc-montpellier.fr
The changes that have been made are:
- Use a different skin for the back-office interface.
- Custom front-end interface.
- Override the Service class.
- Override the Authentication class.
Please note that anyone can login the back-office administration interface and create new services. Thus, for security reasons, we intentionally deactivated the jobs running procedure.
WAVES Singularity image
This is a Singularity container with a functional WAVES installation including two pre-configured services (‘Hello world’ and ‘PhyML’). For testing purpose.
- Singularity image: http://old.atgc-montpellier.fr/download/binaries/waves/wavestest.simg
- Documentation: http://waves-core.readthedocs.io/en/latest/extensions.html#just-have-a-try
Contact
You may contact the WAVES support by e-mail : waves [at] lirmm.fr

Other tools
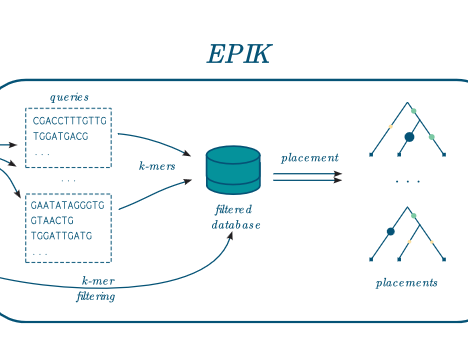
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…
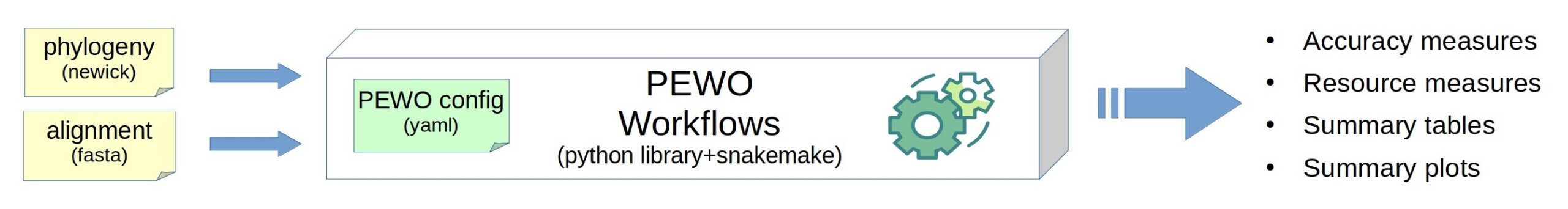
PEWO: a collection of workflows…
Introduction and context In the Bioinformatics team of the LIRMM (CNRS & Univ. Montpellier), we develop a series of tools for metagenomics / metabarcoding analysis. Our tools exploit phylo-k-mers (which are k-mers combined with phylogenetic information) computed for an input set of reference sequences and their phylogeny. The phylo-k-mers are computed and indexed with IPK,…