CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies
CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared collections. The platform is flexible enough to allow each user to focus on some trees or to follow trees manipulated by another user. The platform offers various functionalities covering tree edition, tree comparison, supertree inference, data management.
Why CompPhy?
Online collaborative and real-time tools are needed in various domains for projects involving remote partners. In the last twenty years, web technologies have enabled the development of such tools to jointly edit office documents. However, only few such tools exist in scientific domains. Particularly in phylogentics, prior to the introduction of the CompPhy website, no collaborative tool was available to handle phylogenies.
Key features
- Collaborative editing collection of trees: shared tree visualization, (a)synchronous manipulation of trees, data exchange/storage, …
- Tree edition and taxa annotation: coloration and fonts, shape of the tree, subtree collapsing, …
- Tree comparison: side-by-side display, swapping leaves to ease comparison, MAST, Robinson & Foulds distance
- Supertree inference: MRP, Physic_IST, Astral
- Data management: timeline, associated documents, message forum, backups, …
Access the tool
Use CompPhy here: http://old.atgc-montpellier.fr/compphy/
User guide
Consult the user guide to discover all functionalities.
Scientific reference
« CompPhy: a web-based Collaborative Platform for Comparing Phylogenies », N. Fiorini, V. Lefort, F. Chevenet, V. Berry, A.-M. Arigon Chifolleau, BMC Evol Biol. 2014 Dec 14; 14(1):253.

Other tools
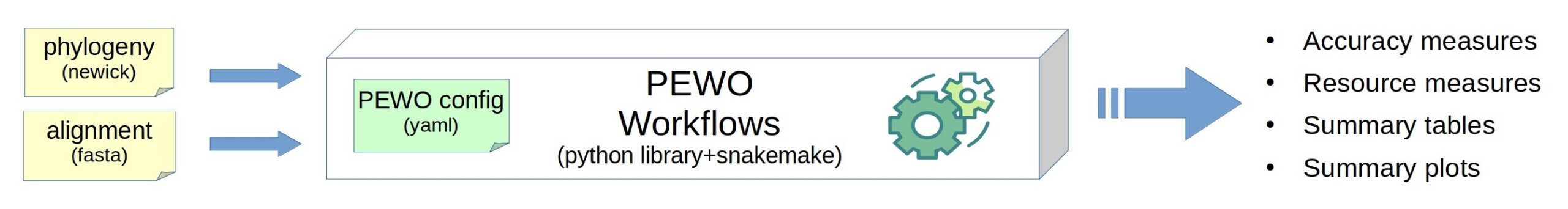
PEWO: a collection of workflows…
Introduction and context In the Bioinformatics team of the LIRMM (CNRS & Univ. Montpellier), we develop a series of tools for metagenomics / metabarcoding analysis. Our tools exploit phylo-k-mers (which are k-mers combined with phylogenetic information) computed for an input set of reference sequences and their phylogeny. The phylo-k-mers are computed and indexed with IPK,…

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
PhyML 3.0
Overview: new algorithms, methods and utilities PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient…