TFscope
Characterizing the binding preferences of transcription factors (TFs) in different cell types and conditions is key to understand how they orchestrate gene expression. TFscope is a machine learning approach that identifies sequence features explaining the binding differences observed between two ChIP-seq experiments targeting either the same TF in two conditions or two TFs with similar motifs (paralogous TFs). TFscope systematically investigates differences in the core motif, nucleotide environment and co-factor motifs, and provides the contribution of each key feature in the two experiments.
TFscope online execution

Other tools
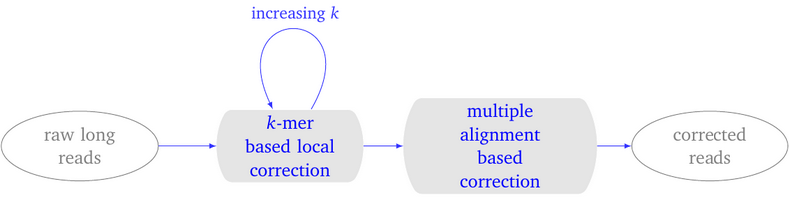
LoRMA: a self correction program…
Overview LoRMA is an error correction program for long reads, which are sequences obtained using the third generation of sequencing technologies (3GS), either with Oxford Nanopore technology or with Pacific Biosciences technology. LoRMA is a so-called self-correction software, as opposed to e.g. LoRDEC that is a hybrid error correction tool. This means that LoRMA uses…

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…
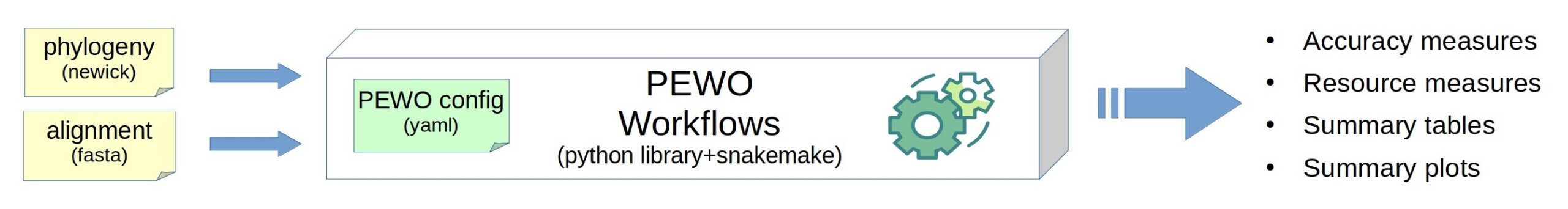
PEWO: a collection of workflows…
Introduction and context In the Bioinformatics team of the LIRMM (CNRS & Univ. Montpellier), we develop a series of tools for metagenomics / metabarcoding analysis. Our tools exploit phylo-k-mers (which are k-mers combined with phylogenetic information) computed for an input set of reference sequences and their phylogeny. The phylo-k-mers are computed and indexed with IPK,…